ABSTRACT
Objective:
Initial diagnosis of medullary thyroid carcinoma (MTC) is frequently associated with advanced stages and a poor prognosis. Thus, the need for earlier diagnoses and detection in relatives at risk for the disease has led to increased use of RET genetic screening.
Subjects and methods:
We performed RET screening in 247 subjects who were referred to the Brazilian Research Consortium for Multiple Endocrine Neoplasia (BRASMEN) Center in the State of Ceará. Direct genetic sequencing was used to analyze exons 8, 10, 11, and 13-16 in MTC index cases and specific exons in at risk relatives. Afterward, clinical follow-up was offered to all the patients with MTC and their affected relatives.
Results:
RET screening was performed in 60 MTC index patients and 187 at-risk family members. At the initial clinical assessment of the index patients, 54 (90%) were diagnosed with apparently sporadic disease and 6 (10%) diagnosed with hereditary disease. After RET screening, we found that 31 (52%) index patients had sporadic disease, and 29 (48%) had hereditary disease. Regarding at-risk relatives, 73/187 were mutation carriers. Mutations in RET codon 804 and the rare p.M918V mutation were the most prevalent.
Conclusions:
Performing RET screening in Ceará allowed us to identify a different mutation profile in this region compared with other areas. RET screening also enabled the diagnosis of a significant number of hereditary MTC patients who were initially classified as sporadic disease patients and benefited their relatives, who were unaware of the risks and the consequences of bearing a RET mutation.
Keywords
Medullary thyroid carcinoma; RET mutation; genetic screening
INTRODUCTION
Medullary thyroid carcinoma (MTC) is an uncommon malignant tumor arising from the calcitonin-producing parafollicular cells (C-cells) of the thyroid and accounts for 1-2% of all thyroid cancers in the US (11. Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567-610.). Regarding the Brazilian population, the latest estimates indicate that 5,870 and 1,090 new cases of all types of thyroid cancer have been diagnosed among women and men, respectively, in 2016 (22. Instituto Nacional de Câncer José Alencar Gomes da Silva. Estimativa 2016: Incidência de Câncer no Brasil. CdPeV, editor. Rio de Janeiro: INCA; 2015. 122 p.). Regarding Ceará, a State of the Northeastern region of Brazil with an estimated population of 9,107,101 inhabitants, the latest estimates indicate that 460 and 100 new cases of all types of thyroid cancer have been diagnosed among women and men, respectively (22. Instituto Nacional de Câncer José Alencar Gomes da Silva. Estimativa 2016: Incidência de Câncer no Brasil. CdPeV, editor. Rio de Janeiro: INCA; 2015. 122 p.).
In a Brazilian study about thyroid cancer incidence patterns in the State of São Paulo from 1997-2008, Veiga and cols. noted incidences of MTC of 0.56 per 100,000 persons in women and 0.15 per 100,000 persons in men (33. Veiga LH, Neta G, Aschebrook-Kilfoy B, Ron E, Devesa SS. Thyroid cancer incidence patterns in Sao Paulo, Brazil, and the U.S. SEER program, 1997-2008. Thyroid. 2013;23:748-57.). Assuming that the incidence of this neoplasm in Ceará is similar to that reported in the State of São Paulo, approximately 26 and 7 new cases of MTC would be diagnosed in women and men, respectively, per year (33. Veiga LH, Neta G, Aschebrook-Kilfoy B, Ron E, Devesa SS. Thyroid cancer incidence patterns in Sao Paulo, Brazil, and the U.S. SEER program, 1997-2008. Thyroid. 2013;23:748-57.).
The sporadic form of MTC is the most common presentation of MTC (approximately 75% of cases) (44. Wells SA Jr, Pacini F, Robinson BG, Santoro M. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab. 2013;98:3149-64.), but in 25% of cases, MTC may also occur as part of an inherited disorder referred to as multiple endocrine neoplasia type 2 (MEN 2). MEN 2 is caused by germline mutations in the REarranged during Transfection (RET) gene and is transmitted as an autosomal dominant trait with variable degrees of expressivity and age-related penetrance (55. Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92:4725-9.).
The clinical diagnosis of MTC by palpation of a thyroid nodule or mass on physical examination is usually associated with advanced TNM stages and a poor prognosis. In this context, RET molecular analysis assumes vital importance, as this analysis allows earlier diagnosis of MTC, thereby increasing the chance of curative treatment (11. Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567-610.). Additionally, RET molecular analysis enables the diagnosis of hereditary MTC in some apparently sporadic cases, which favors more careful management of other diseases that may accompany MEN 2 syndrome.
This study aimed to report the clinical pattern of MTC in the State of Ceará and to describe the molecular profile of this neoplasm using RET gene analysis.
SUBJECTS AND METHODS
Subjects
We report the results of our experience with RET genetic screening of patients with a diagnosis of MTC, as well as their at-risk relatives, who were evaluated at a single center in Ceará, Brazil, from May 2009 to December 2015. This center is located in the city of Fortaleza, the capital of Ceará, and is part of a Brazilian Research Consortium for Multiple Endocrine Neoplasia (BRASMEN). All RET sequencings performed in this study were conducted in the Laboratory of Molecular and Translational Endocrinology, Division of Endocrinology, Department of Medicine, Escola Paulista de Medicina, Universidade Federal de São Paulo (EPM/Unifesp).
All subjects investigated in this study signed informed consent. The study protocol was approved by the local internal review board (CAAE: 16441414.9.1001.5505111).
The patients included in this study had histopathologically confirmed diagnoses of MTC. Immunohistochemical studies for calcitonin, synaptophysin, and chromogranin were performed when histopathological exams were inconclusive, and no patients presented with elevated serum calcitonin (sCt) or CEA levels. All the patients were originally from Ceará. The exclusion criteria were: patients from other countries or other Brazilian States, relatives of deceased MTC index patients without available histopathologic information to confirm their relative's MTC diagnosis and patients with uncertain histopathology.
Clinical evaluation before RET screening
Patients initially underwent a clinical assessment and genetic counseling before undergoing peripheral blood collection for RET analysis. Patients were warned about the risks of having additional endocrine diseases related to MEN 2 if a RET mutation was identified, as well as the risks facing their relatives bearing the same mutation. Therefore, a portion of the patient population analyzed in this study comprised the at-risk relatives of patients diagnosed with hereditary MTC after RET sequencing.
In addition, a questionnaire was designed to acquire the following general information about each patient: age upon diagnosis of MTC/CCH and upon RET screening; previous knowledge of MTC/CCH upon total thyroidectomy (TT), if previously performed; preoperative cervical ultrasound or thyroid nodule or lymph node (LN) fine-needle aspiration biopsy (FNAB) findings; sCt levels; and the referring doctor's specialty.
RET mutation analysis
Genomic DNA was extracted from peripheral blood leucocytes using an in-house protocol (66. Kizys MM, Cardoso MG, Lindsey SC, Harada MY, Soares FA, Melo MC, et al. Optimizing nucleic acid extraction from thyroid fine-needle aspiration cells in stained slides, formalin-fixed/paraffin-embedded tissues, and long-term stored blood samples. Arq Bras Endocrinol Metabol. 2012;56:618-26.). Sequence analysis of hot-spot-bearing exons 8, 10, 11, and 13-16 was performed, and extended RET gene analysis was also conducted (77. Lindsey SC, Kunii IS, Germano-Neto F, Sittoni MY, Camacho CP, Valente FO, et al. Extended RET gene analysis in patients with apparently sporadic medullary thyroid cancer: clinical benefits and cost. Horm Cancer. 2012;3:181-6.) in all patients considered to have sporadic disease according to hot-spot exon 8, 10, 11, 13-16 analysis who presented with multifocal MTC or were young and experienced an unfavorable follow-up course (77. Lindsey SC, Kunii IS, Germano-Neto F, Sittoni MY, Camacho CP, Valente FO, et al. Extended RET gene analysis in patients with apparently sporadic medullary thyroid cancer: clinical benefits and cost. Horm Cancer. 2012;3:181-6.).
Clinical evaluation and follow-up after RET screening
In agreement with their physicians, patients with a diagnosis of MTC and the mutation-carrier relatives were subjected to further biochemical and imaging evaluations. Some of the patients continued to be followed up in the Ceará BRASMEN Center.
Patients with MTC who had already undergone surgical treatment at the time of RET genetic screening were initially followed with sCt, CEA, ionized calcium, TSH, and cervical US assessments every 4 months. Imaging procedures to detect metastases, such as abdominal magnetic resonance (MRI) or 3-phase contrast-enhanced multidetector liver CT, thoracic CT, column and sacral MRI and bone scans, were performed whenever sCt levels were higher than 150 pg/mL. Patients with hereditary MTC and their mutation-carrier relatives who had already undergone surgical treatment were followed with the same complementary tests, as well as annual 24-hour urine catecholamine and metanephrine assessments. Patients who were diagnosed with MTC after RET screening were encouraged to undergo specific surgical treatments according to their preoperative sCt and imaging results, and mutation carriers were encouraged to undergo additional evaluations to screen for PHEO, HPTH, and other MEN 2 syndrome components.
Histopathology
The histological diagnosis of MTC was rendered according to the 2015 ATA guidelines (11. Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567-610.), according to the presence of the basic histological pattern of the disease (i.e., typically round, polyhedral, or spindle-shaped and formed sheets or nests, with peripheral palisading in the vascular stroma, and the presence of stromal amyloid) and the typical immunohistochemical (cells positive for calcitonin, synaptophysin and chromogranin) findings.
STATISTICAL ANALYSIS
The results are presented as the mean ± standard deviation (SD) and as ranges (minimum-maximum). Mann-Whitney U tests and analyses of variance (ANOVA) were used to compare quantitative variables between two and three or more groups, respectively, and Fisher's exact test was used to analyze changes in categorical variables. SPSS software (version 23; SPSS Inc., Chicago, IL, USA) was used to perform the analysis. Statistical significance was indicated by p < 0.05.
RESULTS
Patients and relatives
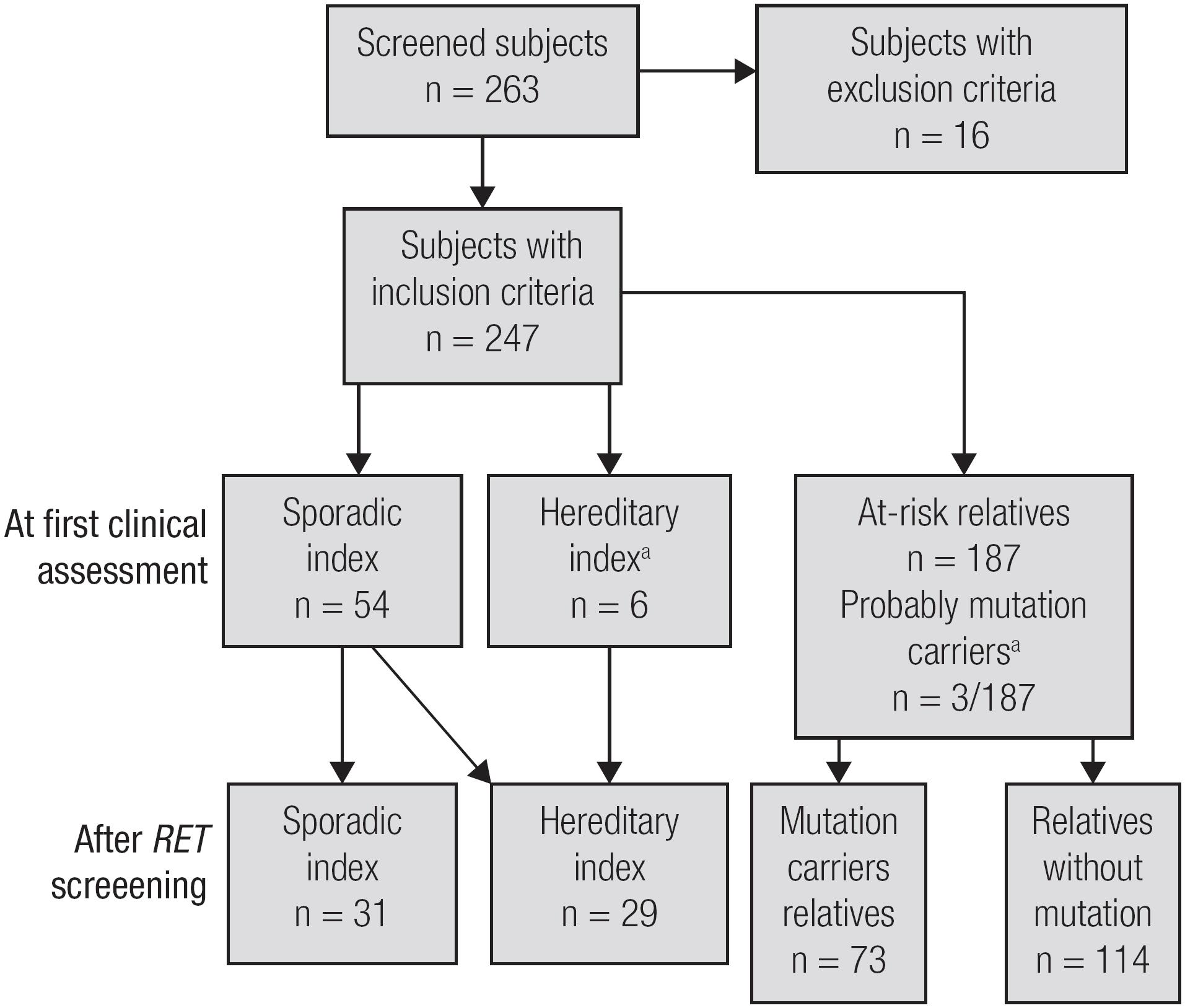
Overall, over the last 5 years, we performed 263 RET sequencing tests in the BRASMEN-Ceará study. Sixteen patients met one or more of the exclusion criteria described above. Therefore, we performed RET genetic screening in 247 subjects, including 60 index patients with MTC and 187 at-risk relatives, from the State of Ceará, Brazil. Head-and-neck surgeons referred most of the index patients (Table 1).
Preoperative clinical data and RET screening results for the index patients with sporadic and hereditary disease and RET mutation-carrier relatives with confirmed diagnoses of MTC/CCH after total thyroidectomy
During the initial phase of the study, based only on RET-screened patient clinical history and physical examination results, we identified only 6 index patients and three relatives with hereditary MTC. These patients were characterized as hereditary index patients because they had another family member with MTC or a personal/familial history of MEN 2. In the second phase of the study, after performing RET sequencing, we observed that 23 MTC index patients who were initially classified as sporadic disease patients were, in fact, hereditary index patients. Regarding at-risk RET-screened relatives, we found that 73 relatives carried mutations, and 114 relatives were non-carriers (Figure 1 and Table 2). The clinical and genetic data of the MTC index patients and the mutation-carrier relatives are shown in Table 2.
Fluxogram of the study according to the first clinical assessment or RET screening. a a This column refers only to relatives who underwent RET screening. Relatives who probably carry RET mutations (by clinical assessment only) but did not undergo RET screening were not included. Diagnoses of MTC/CCH were made based on histopathology results after TT (n = 18) or when relatives who had not undergone TT had fine-needle aspiration cytology positive for MTC concurrently with elevated serum calcitonin levels (above 100 pg/mL) (n = 1). Probable RET mutation carriers identified before genetic screening due to either a personal history of MEN 2 or physical examination findings suggestive of MEN 2.
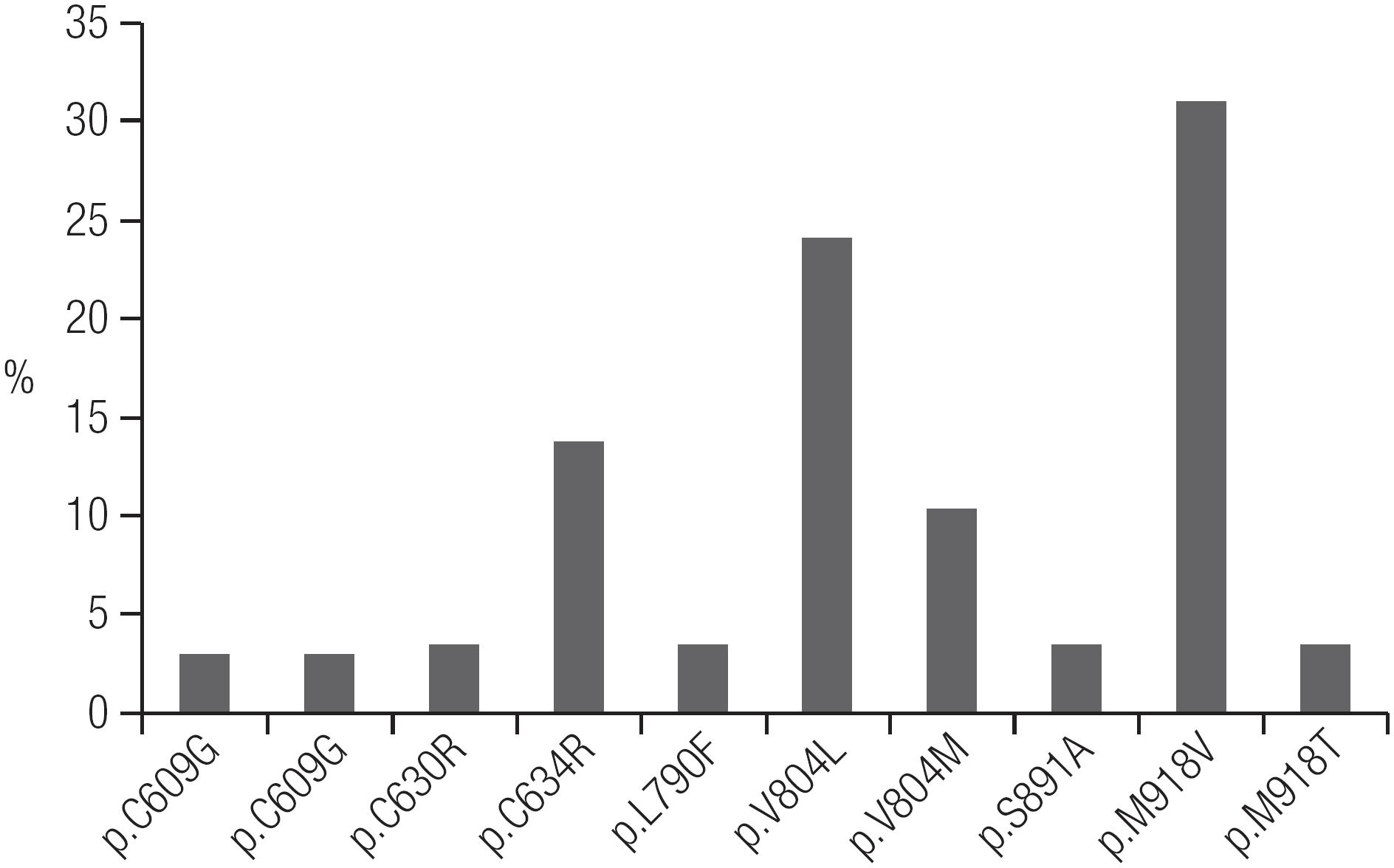
The mutation profile of RET mutations found in Ceará are shown in Figure 2. Overall, of the 29 hereditary MTC index patients who underwent RET screening, 28 definitely underwent TT, and the remaining patient was lost to follow-up (Table 2). In all but one of the MTC index patients, RET screening was performed only postoperatively (Table 1).
Regarding the mutation-carrier relatives, 25 patients presented MTC/CCH confirmed by either histopathology after TT (n = 24) or by FNAB cytology positive for MTC along with elevated sCt levels above 100 pg/mL (n = 1) (Table 2). Among those 24 patients with positive histopathology, 6 had already been diagnosed with MTC before RET screening, as they underwent evaluations for asymptomatic thyroid nodules because they knew about index cases in their families (2 relatives of index patient #4, 1 relative of index patient #12) or because they noticed thyroid nodule growth but were not aware that they had a family member with MTC until a broad pedigree was performed (3 relatives of index patient #20, Table 2). In the remaining patients (n = 18), RET screening led to the diagnosis of MTC/CCH (Tables 1 and 2). In 2 of these 18 patients, we found only CCH. Only one mutation-carrier relative referred to TT had neither MTC nor CCH on histopathology. She was a p.V804L mutation carrier and had undergone TT at 9 years of age.
Genotype-phenotype correlation
Based on the clinical evaluation and RET genetic screening results, we identified 29 index patients with hereditary MTC and 31 index patients with sporadic MTC (Figure 1). The most frequent mutations found were p.V804L and p.M918V (Table 2, Figure 2). PHEO was associated with the following mutations: p.C634R, p.V804L, and p.M918T. HPTH was associated with p.C634R and p.V804L, and LCA was associated with p.C634R. There were no cases of MEN 2A with HD (Table 2). Patients with the p.M918B mutation did not present the MEN 2B phenotype (88. Martins-Costa MC, Cunha LL, Lindsey SC, Camacho CP, Dotto RP, Furuzawa GK, et al. M918V RET mutation causes familial medullary thyroid carcinoma: study of 8 affected kindreds. Endocr Relat Cancer. 2016;23:909-20.). We identified only one patient with MEN 2B, who had the classical p.M918V mutation.
Unsuspected germline RET mutations were found in 23 of the 54 MTC patients (43%) who presented with apparent sporadic disease, according to their negative familial histories and MTC-only phenotypes. These patients were informed about the hereditary origins of their neoplasms, and RET genetic screening was subsequently offered to their relatives (Figure 1).
Preoperative evaluation of RET-screened patients
The preoperative data of the RET-screened patients who underwent TT are shown in Table 1. The majority of patients were women. Additionally, the mutation-carrier relatives whose neoplasm diagnoses were made after RET screening were younger than the patients whose neoplasm diagnoses were made before genetic analysis (not significant).
In most patients, MTC/CCH was diagnosed based on histopathological findings following TT because only 20% of patients had cytology results suggestive of MTC (Figure 3), and consequently, only a few patients had preoperative sCt measurements. However, as expected, we observed lower levels of sCt in mutation-carrier relatives than in hereditary index patients, although the difference in tumor marker levels between the two groups was not statistically significant (Table 1).
Preoperative cytology of index patients with sporadic and hereditary disease and their RET mutation-carrier relatives.
In 9 of the 18 mutation-carrier relatives diagnosed with MTC/CCH after RET screening, diagnoses for MTC had already been made possible by their clinical evaluations, as they had sCt levels above 100 pg/mL along with a suspicious thyroid nodule on US and/or FNAB thyroid nodule or cervical LN cytology results that were suspicious for malignancy (Table 1).
Postoperative evaluation
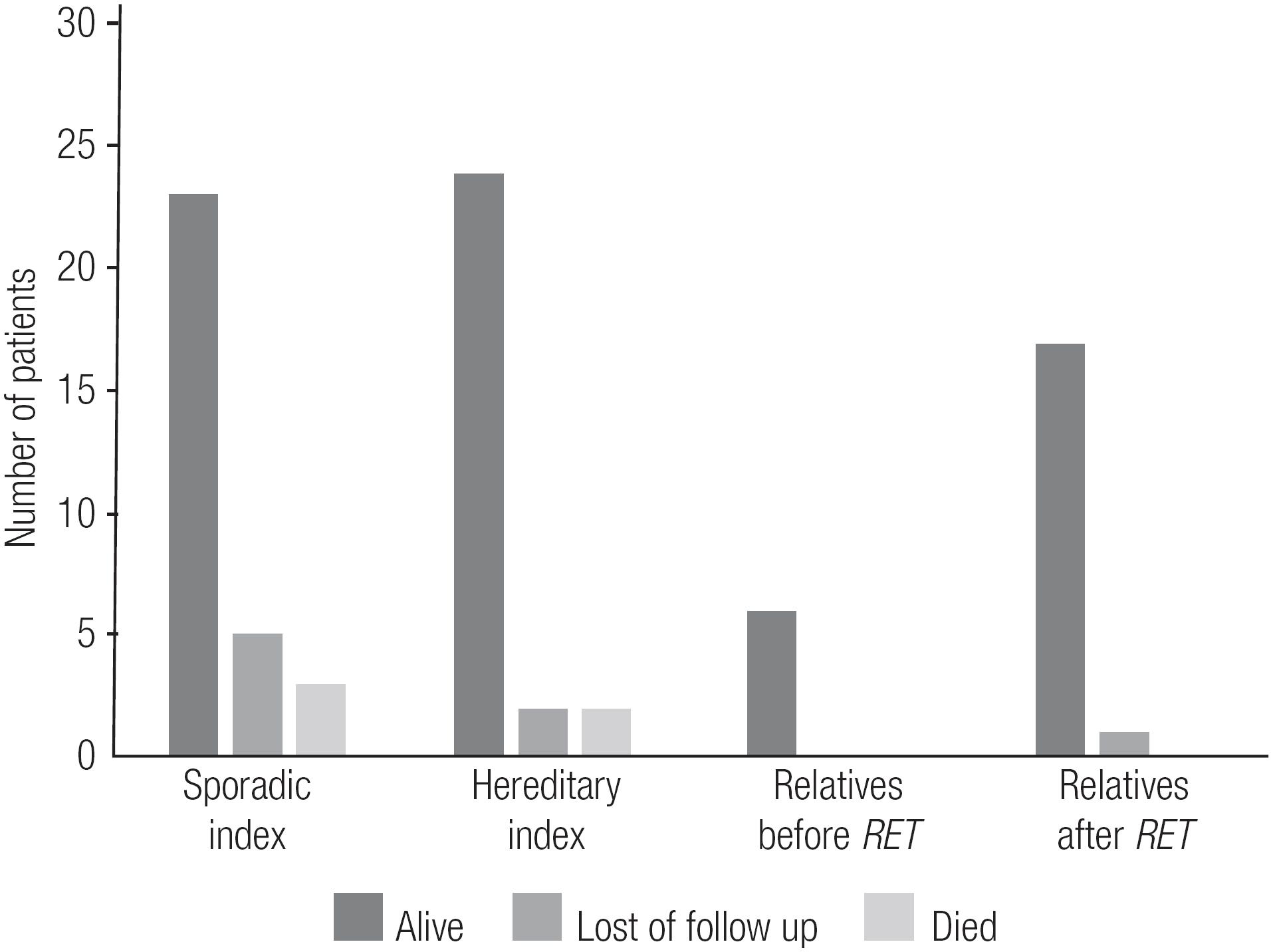
The postoperative data for the RET-screened patients who underwent TT and received a confirmed diagnosis of MTC are shown in Tables 3 and 4. An interesting finding was that relatives whose diagnoses of MTC/CCH were made after RET screening had lower sCt levels and less frequent postsurgical hypoparathyroidism than index patients and mutation-carrier relatives whose diagnoses of MTC/CCH were made before RET screening (p < 0.05) (Table 3). Also, up to the most current follow-up, all relatives diagnosed with MTC/CCH after RET screening were alive, while 5 index cases had died (Figure 4, Table 3).
Current follow-up status of index (sporadic and hereditary) patients and RET-carriers relatives who underwent total thyroidectomy.
Postoperative clinical data for the index patients with sporadic and hereditary disease and RET mutation-carrier relatives with confirmed MTC/CCH after total thyroidectomy
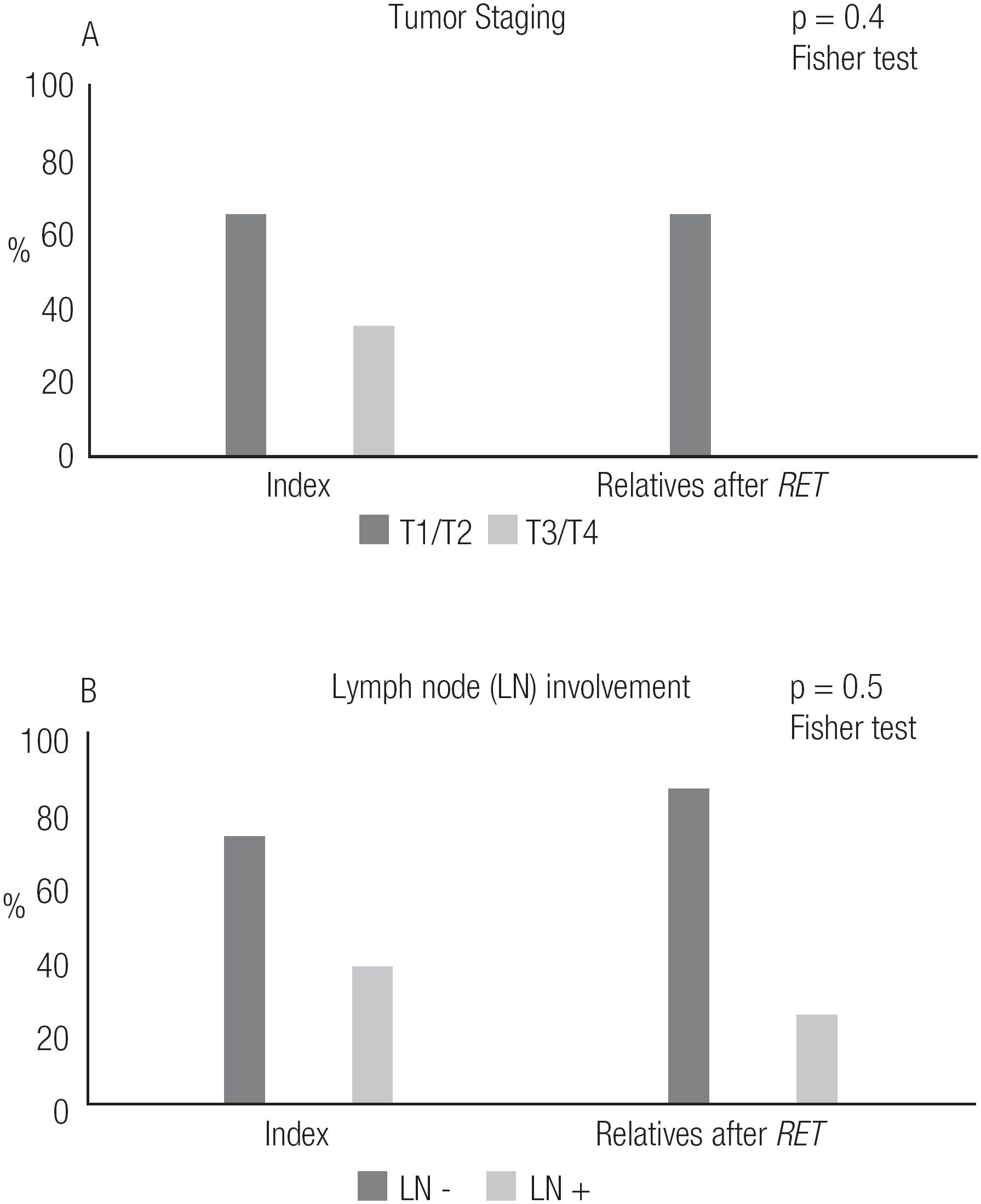
Regarding pathologic tumor and LN staging, we observed that relatives whose diagnoses of MTC/CCH were made after RET screening had smaller tumor dimensions than index patients (p < 0.05), although there was no significant difference in the frequency of LN involvement between the two groups (Figure 5, Table 4).
Postoperative staging of the index patients with sporadic and hereditary disease and RET mutation-carrier relatives.
Postoperative staging of the index patients with sporadic and hereditary disease and RET mutation-carrier relatives with histopathologically confirmed MTC/CCH after total thyroidectomy
A higher proportion of index patients with hereditary disease and their relatives presented tumor multifocality than did index patients with sporadic disease, although the percentage of multifocality was not negligible (22%) in the latter group (Table 4). This finding led us to perform extended RET gene analysis in index patients with sporadic disease to exclude the presence of mutations in other exons. However, no patients presented with new mutations in extended RET gene analysis.
We observed simultaneous MTC and papillary thyroid carcinoma (PTC) in 9 of 78 (11.5%) patients with MTC (Table 4). In some patients, this association could not be evaluated because these patients had previously undergone a partial thyroidectomy and did not have available histopathology results.
DISCUSSION
Identification of RET mutations in patients with a diagnosis of MTC is essential, both for index patients and for at-risk relatives, and allows for the potential identification of hereditary MTC patients appearing to have sporadic MTC who are not followed-up regarding other MEN 2A components, especially the life-threatening disease PHEO (11. Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567-610.). For patients who are already clinically suspected of having hereditary MTC, this analysis allows identification of the specific RET mutation and an estimation of the disease's likely clinical course. For affected family members, RET mutation analysis allows MTC to be cured when prophylactic thyroidectomy is performed and also allows precocious detection of and administration of therapy for other components of the syndrome. For non-affected family members, it spares these patients from having to undergo clinical and biochemical monitoring for all MEN 2A components (99. Frank-Raue K, Raue F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res. 2015;204:139-56.).
Using clinical history and physical examination findings alone, we identified only 6 hereditary MTC index cases. However, RET screening allowed us to identify unsuspected germline RET mutations in 23 of the 54 MTC patients (43%) who appeared to present as sporadic disease cases, according to their negative family histories and isolated MTC phenotype (Table 2). Therefore, through RET screening, the number of index patients with hereditary MTC increased to 29. Elisei and cols. also found unsuspected germline RET mutations in some sporadic cases but at a percentage (only 7.3% of cases) that was much lower than in our study (55. Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92:4725-9.). We believe that some particular findings of this study may explain this difference, such as the higher prevalence of the p.M918V mutation (> 30%) among hereditary MTC cases (Figure 2) marked by the isolated MTC phenotype, as well as the higher percentage of hereditary index patients (48%) in this study compared to the 25% usually described in the literature (55. Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92:4725-9.,1010. Maia AL, Siqueira DR, Kulcsar MA, Tincani AJ, Mazeto GM, Maciel LM. Diagnosis, treatment, and follow-up of medullary thyroid carcinoma: recommendations by the Thyroid Department of the Brazilian Society of Endocrinology and Metabolism. Arq Bras Endocrinol Metabol. 2014;58:667-700.
11. Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19:565-612.-1212. Pacini F, Castagna MG, Brilli L, Pentheroudakis G. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 Suppl 5:v214-9.). Regarding this higher percentage of hereditary index patients in Ceará compared to the literature, we believe that this may reflect the high frequency of mutation dispersion in the past, before the existence of birth control policies. Families were larger and comprised many children. In addition, the frequency of extramarital relationships, which has been reported to be a prevalent practice, was higher and may have resulted in an increased number of births outside of marriage (1313. Freyre G. Casa Grande e Senzala. Edição comemorativa 80 anos ed. Rio de Janeiro: Global; 2012. 728 p.).
In our initial evaluation, the most common mutation found in Ceará was p.M918V (Figure 2). Thus far, this mutation has been identified in 9 families who were apparently unrelated. Previously, this mutation had been described in only one patient with MTC and in 1 family member without a neoplasm in the literature (1414. Cosci B, Vivaldi A, Romei C, Gemignani F, Landi S, Ciampi R, et al. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr Relat Cancer. 2011;18:603-12.). As the number of index patients who were found to have the p.M918V mutation was increased, we observed that some of those patients lived in nearby regions, such as in the Northeastern region of Ceará State. This finding led us to question whether a founder effect could explain the high frequency of the p.M918V mutation in Ceará, as at that time, 8 families with a p.M918V mutation had already been identified. The evidence suggestive of a founder effect as an explanation for the frequency of the p.M918V mutation in Ceará led us to consider whether the first 8 families diagnosed with a p.M918V mutation were actually one large family (88. Martins-Costa MC, Cunha LL, Lindsey SC, Camacho CP, Dotto RP, Furuzawa GK, et al. M918V RET mutation causes familial medullary thyroid carcinoma: study of 8 affected kindreds. Endocr Relat Cancer. 2016;23:909-20.).
The most frequent mutations identified in our study were in codons 918 and 804 (Figure 2, Table 2), findings that differ from those of other studies worldwide, in which mutations in codon 634 were the most common mutations found (1515. Sanso GE, Domene HM, Garcia R, Pusiol E, de M, Roque M, et al. Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer. 2002;94:323-30.
16. Alvandi E, Akrami SM, Chiani M, Hedayati M, Nayer BN, Tehrani MR, et al. Molecular analysis of the RET proto-oncogene key exons in patients with medullary thyroid carcinoma: a comprehensive study of the Iranian population. Thyroid. 2011;21:373-82.
17. Sharma BP, Saranath D. RET gene mutations and polymorphisms in medullary thyroid carcinomas in Indian patients. J Biosci. 2011;36:603-11.
18. Chung YJ, Kim HH, Kim HJ, Min YK, Lee MS, Lee MK, et al. RET proto-oncogene mutations are restricted to codon 634 and 618 in Korean families with multiple endocrine neoplasia 2A. Thyroid. 2004;14:813-8.
19. Zhou Y, Zhao Y, Cui B, Gu L, Zhu S, Li J, et al. RET proto-oncogene mutations are restricted to codons 634 and 918 in mainland Chinese families with MEN2A and MEN2B. Clin Endocrinol (Oxf). 2007;67:570-6.
20. Qi XP, Chen XL, Ma JM, Du ZF, Fei J, Yang CP, et al. RET proto-oncogene genetic screening of families with multiple endocrine neoplasia type 2 optimizes diagnostic and clinical management in China. Thyroid. 2012;22:1257-65.
21. Niccoli-Sire P, Murat A, Rohmer V, Franc S, Chabrier G, Baldet L, et al. Familial medullary thyroid carcinoma with noncysteine ret mutations: phenotype-genotype relationship in a large series of patients. J Clin Endocrinol Metab. 2001;86:3746-53.
22. Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol. 2010;163:301-8.
23. Nguyen L, Niccoli-Sire P, Caron P, Bastie D, Maes B, Chabrier G, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. Eur J Endocrinol. 2001;144:37-44.
24. Machens A, Niccoli-Sire P, Hoegel J, Frank-Raue K, van Vroonhoven TJ, Roeher HD, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349:1517-25.
25. Machens A, Lorenz K, Sekulla C, Hoppner W, Frank-Raue K, Raue F, et al. Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol. 2013;168:307-14.
26. Toledo RA, Hatakana R, Lourenco DM Jr., Lindsey SC, Camacho CP, Almeida M, et al. Comprehensive assessment of the disputed RET Y791F variant shows no association with medullary thyroid carcinoma susceptibility. Endocr Relat Cancer. 2015;22:65-76.-2727. Toledo RA, Maciel RM, Erlic Z, Lourenco DM Jr., Cerutti JM, Eng C, et al. RET Y791F Variant Does Not Increase the Risk for Medullary Thyroid Carcinoma. Thyroid. 2015;25:973-4.). Also, mutations in codons 338, 515, 666, 618, 620, 768, 848, 883 and 904, which were observed in the European studies (2525. Machens A, Lorenz K, Sekulla C, Hoppner W, Frank-Raue K, Raue F, et al. Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol. 2013;168:307-14.), have not been identified in Ceará thus far (Table 5).
Prevalence of RET gene mutations in the State of Ceará along with major international studies and their respective genotype-phenotype correlations
Regarding preoperative evaluations, the low sensitivity of FNAB cytology noted in this study (20%) (Figure 3) had previously been reported in other studies (2828. Essig GF Jr, Porter K, Schneider D, Debora A, Lindsey SC, Busonero G, et al. Fine needle aspiration and medullary thyroid carcinoma: the risk of inadequate preoperative evaluation and initial surgery when relying upon FNAB cytology alone. Endocr Pract. 2013;19:920-7.
29. Bugalho MJ, Santos JR, Sobrinho L. Preoperative diagnosis of medullary thyroid carcinoma: fine needle aspiration cytology as compared with serum calcitonin measurement. J Surg Oncol. 2005;91:56-60.-3030. Chang TC, Wu SL, Hsiao YL. Medullary thyroid carcinoma: pitfalls in diagnosis by fine needle aspiration cytology and relationship of cytomorphology to RET proto-oncogene mutations. Acta Cytol. 2005;49:477-82.). Essig Jr and cols. concluded that cytological evaluations alone limited one's ability to perform an optimal preoperative assessment and initial surgery in over half of affected patients in sporadic MTC (2828. Essig GF Jr, Porter K, Schneider D, Debora A, Lindsey SC, Busonero G, et al. Fine needle aspiration and medullary thyroid carcinoma: the risk of inadequate preoperative evaluation and initial surgery when relying upon FNAB cytology alone. Endocr Pract. 2013;19:920-7.).
The present study led us to analyze the potential benefits of diagnosing affected relatives. Comparison of mutation-carrier relatives diagnosed with MTC after RET screening to index patients showed that those relatives had a lower frequency of complications, such as post-surgical hypoparathyroidism, and lower levels of sCt upon their last visit (p < 0.05). Also, none of the affected relatives died, while 5 indexes succumbed to disease (Figure 4). Interestingly, the affected relatives were not diagnosed at a significantly younger age (Table 3), nor were they diagnosed at a more favorable clinical or pathologic stage than their counterparts (Table 4). In fact, a trend toward diagnosis at a younger age was observed, but the trend was not statistically significant (Table 3). We also observed that relatives who had a diagnosis of MTC after RET screening had smaller tumor dimensions than index patients, but the degree of lymph node involvement was similar between the two groups (Figure 5). In fact, 50% of those mutation-carrier relatives diagnosed with MTC after RET screening already presented clinical evidence of MTC before the RET screening results were available (Table 1). Therefore, in our study population, the benefits of RET screening were not fully realized. We consider that the major reason for this was because these patients received this exam relatively late, as they were not aware of its existence and benefits. We believe that if we can offer earlier RET screening for at-risk patients, we will be able to diagnose neoplasms at an earlier stage, or, more desirably, before they have developed.
The finding of simultaneous MTC and PTC in this study (11.5%) had previously been described in previous studies and was observed in the following variable percentages of patients in those studies: 3.6% of patients in Germany (3131. Machens A, Dralle H. Simultaneous medullary and papillary thyroid cancer: a novel entity? Ann Surg Oncol. 2012;19:37-44.), 13.8% of patients in Italy (3232. Biscolla RP, Ugolini C, Sculli M, Bottici V, Castagna MG, Romei C, et al. Medullary and papillary tumors are frequently associated in the same thyroid gland without evidence of reciprocal influence in their biologic behavior. Thyroid. 2004;14:946-52.) and 19% of patients in Korea (3333. Kim WG, Gong G, Kim EY, Kim TY, Hong SJ, Kim WB, et al. Concurrent occurrence of medullary thyroid carcinoma and papillary thyroid carcinoma in the same thyroid should be considered as coincidental. Clin Endocrinol (Oxf). 2010;72:256-63.). Those discrepancies were attributed to environmental conditions and differences in the populations of the studies in question (11. Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567-610.).
Regarding tumor multifocality, this phenomenon was observed more frequently in index patients with hereditary disease and their relatives than in index patients with sporadic disease. Consistent with the findings of the study by Lindsey and cols. (77. Lindsey SC, Kunii IS, Germano-Neto F, Sittoni MY, Camacho CP, Valente FO, et al. Extended RET gene analysis in patients with apparently sporadic medullary thyroid cancer: clinical benefits and cost. Horm Cancer. 2012;3:181-6.), in the present study, we noted no direct clinical benefit to extending the RET germline analysis beyond the hot-spot regions in patients with sporadic MTC with multifocality (22%) since the analysis did not identify any mutations in other exons. Therefore, these cases were true sporadic cases of MTC with multifocal tumors, which had been observed in a recent study (3434. Essig GF Jr, Porter K, Schneider D, Arpaia D, Lindsey SC, Busonero G, et al. Multifocality in Sporadic Medullary Thyroid Carcinoma: An International Multicenter Study. Thyroid. 2016;26:1563-72.).
Our study population comprised a significant number of mutation carriers who did not attend follow-up visits after learning their RET sequencing results, nor did they bring their at-risk relatives to a clinic to undergo RET screening. Geographic distances and some patients’ poor financial statuses, which prevented them from bearing the financial burden imposed by traveling to our center, are recognized as important factors that delayed the performance of RET screening. We also considered other reasons that may explain some patients’ delays in undergoing genetic analysis and some patients’ refusal to undergo genetic analysis. First, not every RET gene carrier is emotionally prepared to face a diagnosis of MTC or to share personal genetic information with his or her relatives. These types of behavior may be related to feelings of guilt and resentment, emotional distress, and poor familial interactions (2525. Machens A, Lorenz K, Sekulla C, Hoppner W, Frank-Raue K, Raue F, et al. Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol. 2013;168:307-14.,3535. Machens A, Dralle H. Multiple endocrine neoplasia type 2: achievements and current challenges. Clinics (Sao Paulo). 2012;67 Suppl 1:113-8.). Other obstacles affecting patients’ abilities to undergo genetic analysis are related to individual aspects, such as patients’ understanding of their disease and religious beliefs. In contrast to the above findings, in some families, we observed solidarity among family members. Additionally, we noted that some family members felt a need to reveal their diagnosis and their intention to face their disease positively in order to motivate their relatives to accelerate their diagnostic and treatment processes to avoid being diagnosed with a neoplasm at the same stage that they had been.
In conclusion, in our study, RET screening was profoundly crucial for the early diagnosis of MTC and facilitated monitoring of other MEN 2A components and identification of unsuspected germline mutations in index patients with apparent sporadic disease. We noted a mutation profile that differed from that of previous European studies. We attribute these differences to our having restricted our analysis to the State of Ceará and to our having evaluated a small number of patients. These features make the extrapolation of our results to the rest of the country unlikely, as different colonization patterns occurred throughout Brazil. However, we believe that any regional differences in RET distributions that exist are likely to diminish as a result of dynamic world social, political and economic scenarios impacting migration processes.
-
Disclosure: no potential conflict of interest relevant to this article was reported.
Acknowledgments:
we would like to express our sincere appreciation to the medical team of Ceará (including the endocrinologists, head-and-neck surgeons, general surgeons, oncologists, nuclear medicine physicians, radiotherapists, radiologists, and pathologists), whose cooperation made this work possible. We would like to give our posthumous thanks to Mr. Antônio Adriano Mendes, who kindly collaborated with us to produce the pedigrees. We also thank Ângela Maria Faria for her administrative assistance and the Federal Agency for the Support and Evaluation of Graduate Education (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES), the National Council for Scientific and Technological Development (CNPq) and the São Paulo Research Foundation (Fundação de Amparo à Pesquisa do Estado de São Paulo – Fapesp) for providing financial support.
REFERENCES
-
1Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567-610.
-
2Instituto Nacional de Câncer José Alencar Gomes da Silva. Estimativa 2016: Incidência de Câncer no Brasil. CdPeV, editor. Rio de Janeiro: INCA; 2015. 122 p.
-
3Veiga LH, Neta G, Aschebrook-Kilfoy B, Ron E, Devesa SS. Thyroid cancer incidence patterns in Sao Paulo, Brazil, and the U.S. SEER program, 1997-2008. Thyroid. 2013;23:748-57.
-
4Wells SA Jr, Pacini F, Robinson BG, Santoro M. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab. 2013;98:3149-64.
-
5Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92:4725-9.
-
6Kizys MM, Cardoso MG, Lindsey SC, Harada MY, Soares FA, Melo MC, et al. Optimizing nucleic acid extraction from thyroid fine-needle aspiration cells in stained slides, formalin-fixed/paraffin-embedded tissues, and long-term stored blood samples. Arq Bras Endocrinol Metabol. 2012;56:618-26.
-
7Lindsey SC, Kunii IS, Germano-Neto F, Sittoni MY, Camacho CP, Valente FO, et al. Extended RET gene analysis in patients with apparently sporadic medullary thyroid cancer: clinical benefits and cost. Horm Cancer. 2012;3:181-6.
-
8Martins-Costa MC, Cunha LL, Lindsey SC, Camacho CP, Dotto RP, Furuzawa GK, et al. M918V RET mutation causes familial medullary thyroid carcinoma: study of 8 affected kindreds. Endocr Relat Cancer. 2016;23:909-20.
-
9Frank-Raue K, Raue F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res. 2015;204:139-56.
-
10Maia AL, Siqueira DR, Kulcsar MA, Tincani AJ, Mazeto GM, Maciel LM. Diagnosis, treatment, and follow-up of medullary thyroid carcinoma: recommendations by the Thyroid Department of the Brazilian Society of Endocrinology and Metabolism. Arq Bras Endocrinol Metabol. 2014;58:667-700.
-
11Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19:565-612.
-
12Pacini F, Castagna MG, Brilli L, Pentheroudakis G. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 Suppl 5:v214-9.
-
13Freyre G. Casa Grande e Senzala. Edição comemorativa 80 anos ed. Rio de Janeiro: Global; 2012. 728 p.
-
14Cosci B, Vivaldi A, Romei C, Gemignani F, Landi S, Ciampi R, et al. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr Relat Cancer. 2011;18:603-12.
-
15Sanso GE, Domene HM, Garcia R, Pusiol E, de M, Roque M, et al. Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer. 2002;94:323-30.
-
16Alvandi E, Akrami SM, Chiani M, Hedayati M, Nayer BN, Tehrani MR, et al. Molecular analysis of the RET proto-oncogene key exons in patients with medullary thyroid carcinoma: a comprehensive study of the Iranian population. Thyroid. 2011;21:373-82.
-
17Sharma BP, Saranath D. RET gene mutations and polymorphisms in medullary thyroid carcinomas in Indian patients. J Biosci. 2011;36:603-11.
-
18Chung YJ, Kim HH, Kim HJ, Min YK, Lee MS, Lee MK, et al. RET proto-oncogene mutations are restricted to codon 634 and 618 in Korean families with multiple endocrine neoplasia 2A. Thyroid. 2004;14:813-8.
-
19Zhou Y, Zhao Y, Cui B, Gu L, Zhu S, Li J, et al. RET proto-oncogene mutations are restricted to codons 634 and 918 in mainland Chinese families with MEN2A and MEN2B. Clin Endocrinol (Oxf). 2007;67:570-6.
-
20Qi XP, Chen XL, Ma JM, Du ZF, Fei J, Yang CP, et al. RET proto-oncogene genetic screening of families with multiple endocrine neoplasia type 2 optimizes diagnostic and clinical management in China. Thyroid. 2012;22:1257-65.
-
21Niccoli-Sire P, Murat A, Rohmer V, Franc S, Chabrier G, Baldet L, et al. Familial medullary thyroid carcinoma with noncysteine ret mutations: phenotype-genotype relationship in a large series of patients. J Clin Endocrinol Metab. 2001;86:3746-53.
-
22Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol. 2010;163:301-8.
-
23Nguyen L, Niccoli-Sire P, Caron P, Bastie D, Maes B, Chabrier G, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. Eur J Endocrinol. 2001;144:37-44.
-
24Machens A, Niccoli-Sire P, Hoegel J, Frank-Raue K, van Vroonhoven TJ, Roeher HD, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349:1517-25.
-
25Machens A, Lorenz K, Sekulla C, Hoppner W, Frank-Raue K, Raue F, et al. Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol. 2013;168:307-14.
-
26Toledo RA, Hatakana R, Lourenco DM Jr., Lindsey SC, Camacho CP, Almeida M, et al. Comprehensive assessment of the disputed RET Y791F variant shows no association with medullary thyroid carcinoma susceptibility. Endocr Relat Cancer. 2015;22:65-76.
-
27Toledo RA, Maciel RM, Erlic Z, Lourenco DM Jr., Cerutti JM, Eng C, et al. RET Y791F Variant Does Not Increase the Risk for Medullary Thyroid Carcinoma. Thyroid. 2015;25:973-4.
-
28Essig GF Jr, Porter K, Schneider D, Debora A, Lindsey SC, Busonero G, et al. Fine needle aspiration and medullary thyroid carcinoma: the risk of inadequate preoperative evaluation and initial surgery when relying upon FNAB cytology alone. Endocr Pract. 2013;19:920-7.
-
29Bugalho MJ, Santos JR, Sobrinho L. Preoperative diagnosis of medullary thyroid carcinoma: fine needle aspiration cytology as compared with serum calcitonin measurement. J Surg Oncol. 2005;91:56-60.
-
30Chang TC, Wu SL, Hsiao YL. Medullary thyroid carcinoma: pitfalls in diagnosis by fine needle aspiration cytology and relationship of cytomorphology to RET proto-oncogene mutations. Acta Cytol. 2005;49:477-82.
-
31Machens A, Dralle H. Simultaneous medullary and papillary thyroid cancer: a novel entity? Ann Surg Oncol. 2012;19:37-44.
-
32Biscolla RP, Ugolini C, Sculli M, Bottici V, Castagna MG, Romei C, et al. Medullary and papillary tumors are frequently associated in the same thyroid gland without evidence of reciprocal influence in their biologic behavior. Thyroid. 2004;14:946-52.
-
33Kim WG, Gong G, Kim EY, Kim TY, Hong SJ, Kim WB, et al. Concurrent occurrence of medullary thyroid carcinoma and papillary thyroid carcinoma in the same thyroid should be considered as coincidental. Clin Endocrinol (Oxf). 2010;72:256-63.
-
34Essig GF Jr, Porter K, Schneider D, Arpaia D, Lindsey SC, Busonero G, et al. Multifocality in Sporadic Medullary Thyroid Carcinoma: An International Multicenter Study. Thyroid. 2016;26:1563-72.
-
35Machens A, Dralle H. Multiple endocrine neoplasia type 2: achievements and current challenges. Clinics (Sao Paulo). 2012;67 Suppl 1:113-8.
Publication Dates
-
Publication in this collection
2018
History
-
Received
29 June 2018 -
Accepted
25 Sept 2018