ABSTRACT
The somatotropic axis is the main hormonal regulator of growth. Growth hormone (GH), also known as somatotropin, and insulin-like growth factor 1 (IGF-1) are the key components of the somatotropic axis. This axis has been studied for a long time and the knowledge of how some molecules could promote or impair hormones production and action has been growing over the last decade. The enhancement of large-scale sequencing techniques has expanded the spectrum of known genes and several other candidate genes that could affect the GH-IGF1-bone pathway. To date, defects in more than forty genes were associated with an impairment of the somatotropic axis. These defects can affect from the secretion of GH to the bioavailability and action of IGF-1. Affected patients present a large heterogeneous group of conditions associated with growth retardation. In this review, we focus on the description of the GH-IGF axis genetic defects reported in the last decade. Arch Endocrinol Metab. 2019;63(6):608-17
Growth hormone; short stature; IGF; somatotropic axis
INTRODUCTION
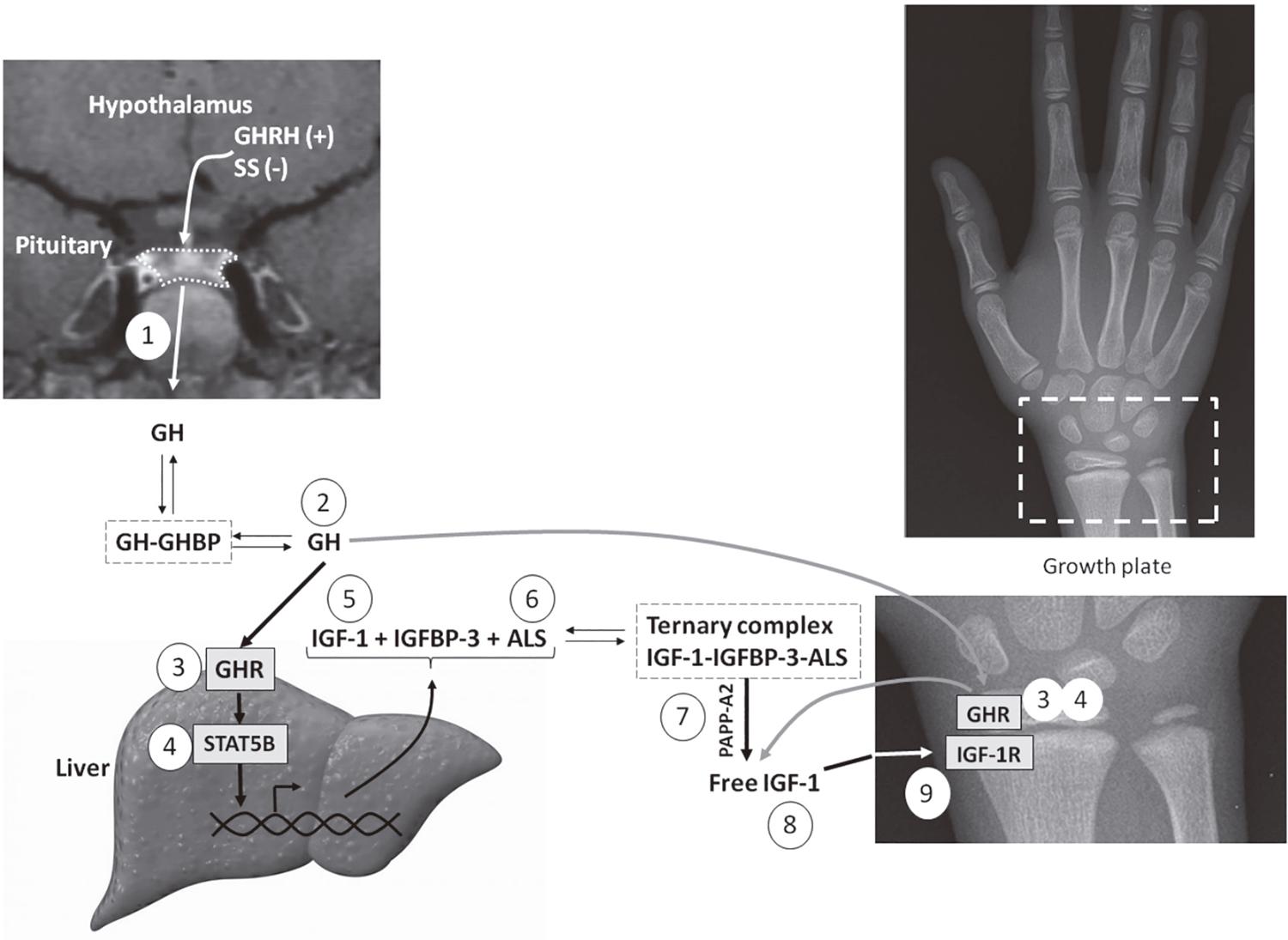
The somatotropic axis is the main hormonal regulator of growth. Growth hormone (GH), also known as somatotropin, and insulin-like growth factor 1 (IGF-11. Ranke MB, Wit JM. Growth hormone – past, present and future. Nat Rev Endocrinol. 2018;14(5):285-300.) are the key components of the somatotropic axis. GH is secreted by somatotropes of the anterior pituitary gland, in a pulsatile manner, under stimulation of growth hormone-releasing hormone (GHRH), produced in the hypothalamus, and ghrelin (GHS), which is also secreted by the gastric cells. Somatostatin, another hypothalamic hormone, has an inhibitory action on GH secretion. GH acts by binding to its receptor (GHR), a homo-dimeric transmembrane receptor belonging to the citokine receptor superfamily. GH binding results in dimerization of the GHR and phosphorylation of JAK2, a tyrosine kinase associated with GHR. JAK2 phosphorylation leads to activation of different intracellular pathways, leading to direct metabolic effects or regulating the gene transcription, which may involve a family of signal transducers and activators of transcription (STATs). The most important STAT implicated in the growth-promoting signaling of GHR is STAT5B. This protein stimulates the synthesis of IGF-1 and of other molecules that increase IGF-1 bioavailability (Figure 1) (11. Ranke MB, Wit JM. Growth hormone – past, present and future. Nat Rev Endocrinol. 2018;14(5):285-300.,22. Waters MJ, Brooks AJ. Growth hormone receptor: structure function relationships. Horm Res Paediatr. 2011;76 Suppl 1:12-6.).
Schematic representation of the GH-IGF axis indicating the main disorders in this system: 1) growth hormone deficiency (GHD); 2) bioinactive GH; 3) GH insensitivity by GHR defects; 4) GH insensitivity by STAT5B defects; 5) IGF-1 deficiency; 6) acid-labile subunit (ALS) deficiency; 7) defects in the proteolytic cleavage of IGFBPs by PAPPA2 deficiency; 8) bioinactive IGF-1; and 9) IGF insensitivity caused by IGF1R defects.
The GH effect of promoting growth occurs mainly through IGF-1 production. Circulating IGF-1 is predominantly produced by the liver. Other tissues can produce IGF-1 that acts in a paracrine/autocrine manner, as occurs in the growth plate. Most of circulating IGF-1 is carried by a ternary complex, consisting of one molecule of IGF-1, one molecule of IGF binding protein type 3 (IGFBP-33. Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180(2):247-55.) and one molecule of acid-labile subunit (ALS). This complex is essential to decrease the availability of free IGF-1 to the tissues, extending its serum half-life (Figure 1). IGF-1 is the main growth factor in postnatal growth. In the prenatal period, IGF-1 and IGF-2 have essential role in intrauterine development. IGF-1and IGF-2 act by binding to the cell surface tyrosine kinase receptor called IGF1R. This binding activates intracellular signaling, as the PI3K/AKT pathway, important for cell growth and proliferation (11. Ranke MB, Wit JM. Growth hormone – past, present and future. Nat Rev Endocrinol. 2018;14(5):285-300.,33. Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180(2):247-55.).
Defects in many genes related to the somatotropic axis lead to a large heterogeneous group of conditions associated with growth retardation (44. Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99(9):3080-92.). Studies in this area are long-standing and, in 1929, researchers hypothesized that some genes would be responsible for pituitary development and function (55. Snell GD. Dwarf, a new Mendelian recessive character of the house mouse. Proc Natl Acad Sci U S A. 1929;15(9):733-4.). After many reports of familial cases of isolated GH deficiency, deletions in GH1 gene were recognized as its first monogenic cause in 1981 (66. Phillips JA, Hjelle BL, Seeburg PH, Zachmann M. Molecular basis for familial isolated growth hormone deficiency. Proc Natl Acad Sci U S A. 1981;78(10):6372-5.). Subsequently, in 1989, autosomal recessive mutations in GHR were established as cause of severe short stature with complete insensitivity to GH (77. Amselem S, Duquesnoy P, Attree O, Novelli G, Bousnina S, Postel-Vinay MC, et al. Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med. 1989;321(15):989-95.). A candidate gene for the hypopituitarism phenotype, named pituitary-specific positive transcription factor 1 (POU1F1, formally PIT1) was reported in 1990 based on a spontaneous dwarf mice (88. Dollé P, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M. Expression of GHF-1 protein in mouse pituitaries correlates both temporally and spatially with the onset of growth hormone gene activity. Cell. 1990;60(5):809-20.). Two years later, an autosomal recessive mutation in POU1F1 was identified in patients with GH, PRL and TSH deficiency (99. Tatsumi K, Miyai K, Notomi T, Kaibe K, Amino N, Mizuno Y, et al. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet. 1992;1(1):56-8.). Also in 90s, inactivating mutations of the transcription factor of POU1F1 gene (PROP1) in an autosomal recessive pattern were also described in individuals with hypopituitarism, with or without gonadotropic deficiency (1010. Correa FA, Nakaguma M, Madeira JLO, Nishi MY, Abrao MG, Jorge AAL, et al. Combined pituitary hormone deficiency caused by PROP1 mutations: update 20 years post-discovery. Arch Endocrinol Metab. 2019;63(2):167-74.). In the same period , homozygous loss-of-function mutations in IGF-1 gene were reported in a child with intrauterine growth retardation, microcephaly, retarded intellectual development, and severe postnatal growth failure (1111. Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335(18):1363-7.). Genes that were associated with GH-IGF-1 axis defects before 2009 are depicted in a historical timeline (Figure 2).
Historical timeline with genes previously associated with GH-IGF-1 axis defects until 2009.
To date, defects in more than forty genes were associated with an impairment of the somatotropic axis. These defects can affect from the secretion of GH to the bioavailability and action of IGF-1. In this review, we describe the GH-IGF axis genetic defects reported in the last ten years. For didactic purpose, we divided this review in two main topics: “new genetic defects associated with GH deficiency” and “new genetic defects associated with GH-IGFs axis”.
NEW GENETIC DEFECTS ASSOCIATED WITH GH DEFICIENCY
Growth hormone deficiency can be either isolated (IGHD) or combined with other pituitary hormones deficiencies (CPHD). It can be congenital or acquired and, regarding neuroimaging, it can present with no abnormalities or a wide range of structural changes of the hypothalamic-pituitary region. The pituitary stalk interruption syndrome (PSIS) has been classified as a distinct clinical picture characterized by hormone deficiencies, a thin, interrupted or absent pituitary stalk, usually associated with an ectopic posterior pituitary and hypoplasia of the anterior pituitary (1212. Pinto G, Netchine I, Sobrier ML, Brunelle F, Souberbielle JC, Brauner R. Pituitary stalk interruption syndrome: a clinical-biological-genetic assessment of its pathogenesis. J Clin Endocrinol Metab. 1997;82(10):3450-4.). Apart from the subtle midline malformation described in the PSIS, it is not unusual that patients with genetic defects present other complex phenotypes. The most common are: complex craniofacial malformations, septo-optic dysplasia (SOD) and developmental delay/intellectual disability (1313. Correa FA, Jorge AA, Nakaguma M, Canton AP, Costa SS, Funari MF, et al. Pathogenic copy number variants in patients with congenital hypopituitarism associated with complex phenotypes. Clin Endocrinol (Oxf). 2018;88(3):425-31.). After the description of the classic and most frequent genetic defects associated with GH deficiency, many other genes have been associated with congenital hypopituitarism, most of them in a small subset of patients with complex phenotypes or PSIS. It has also been recently advocated that some patients could have more than one gene affected, which points to a digenic or oligogenic cause of GH deficiency. However, more studies are needed to explain the individual role of each mutation or its synergy in the pathogenesis of these phenotypes (1414. Zwaveling-Soonawala N, Alders M, Jongejan A, Kovacic L, Duijkers FA, Maas SM, et al. Clues for polygenic inheritance of pituitary stalk interruption syndrome from exome sequencing in 20 patients. J Clin Endocrinol Metab. 2018;103(2):415-28.). Table 1 has a comprehensive list of these new genetic defects, and some of which will be discussed below.
GLI2 – a gene frequently involved in GH deficiency
GLI2 is a transcription factor that contains a zinc-finger region responsible for binding to DNA and is involved in the Sonic Hedgehog (Shh) signaling pathway, which is implicated in cellular proliferation during embryogenesis. It is important for hypothalamus, infundibulum and posterior pituitary lobe formation (1515. Arnhold IJ, Franca MM, Carvalho LR, Mendonca BB, Jorge AA. Role of GLI2 in hypopituitarism phenotype. J Mol Endocrinol. 2015;54(3):R141-50.).
Regarding the clinical picture, patients with GLI2 mutations may have IGHD or CPHD. An ectopic posterior pituitary is a frequent finding as well as polydactyly. In some patients, other midline craniofacial malformations, such as cleft palate and a median maxillary incisor, have been described. The presence of polydactyly should prompt to GLI2 testing and is a marker of severity. There are also rare reports of associated diabetes insipidus. The inheritance is autosomal dominant with incomplete penetration and it is possible that other genetic and/or environmental factors may play a role in the phenotype. In a recent review of the important role of GLI2 mutations in GH deficiency, among 284 patients in whom the complete coding region was sequenced, 15 patients (5%) had either loss-of-function or non-synonymous GLI2 rare variants making it the most frequent genetic defect found in these patients, although one cannot confirm the pathogenicity of all of them (1515. Arnhold IJ, Franca MM, Carvalho LR, Mendonca BB, Jorge AA. Role of GLI2 in hypopituitarism phenotype. J Mol Endocrinol. 2015;54(3):R141-50.,1616. Franca MM, Jorge AA, Carvalho LR, Costalonga EF, Vasques GA, Leite CC, et al. Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J Clin Endocrinol Metab. 2010;95(11):E384-91.).
FGF8, FGFR1, PROKR2 and PROK2 – the overlap between GH deficiency and hypogonadotropic hypogonadism
The hypotheses that mutations in genes underling Kallmann syndrome could also lead to hypopituitarism was made based on the knowledge that an embryogenic structure called preplacodal gives rise to the adenohypophyseal, lens, and olfactory placodes. Therefore, defects in genes involved in the development of these structures could be responsible for both phenotypes (1717. Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012;97(4):E694-9.). Until now, many genes previously associated with hypogonadotropic hypogonadism have also been associated with IGHD or CPHD with or without complex phenotypes. There is a great genetic heterogeneity with autosomal dominant or recessive inheritance with incomplete penetrance. Among the clinical pictures described, there are different combinations of pituitary hormonal deficiencies associated with SOD, HPE, PSIS and Moebius syndrome (1717. Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012;97(4):E694-9.
18. McCabe MJ, Gaston-Massuet C, Tziaferi V, Gregory LC, Alatzoglou KS, Signore M, et al. Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab. 2011;96(10):E1709-18.-1919. Correa FA, Trarbach EB, Tusset C, Latronico AC, Montenegro LR, Carvalho LR, et al. FGFR1 and PROKR2 rare variants found in patients with combined pituitary hormone deficiencies. Endocr Connect. 2015;4(2):100-7.).
ARNT2 and IGSF1 – GH deficiency with very unusual phenotypes
ARNT2 is a transcription factor member of the basic HLH-PAS subfamily proven to be important for hypothalamic development. Using exome sequencing, a mutation in an autosomal mode of inheritance was found in a family with an unusual and distinct combination of clinical manifestations in six affected children (OMIM 615926) (2020. Webb EA, AlMutair A, Kelberman D, Bacchelli C, Chanudet E, Lescai F, et al. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain. 2013;136(Pt 10):3096-105.). The patients had a variable combination of pituitary hormone deficiencies: ADH, ACTH, TSH, LH/FSH and GH. MRI of the brain revealed a similar pattern of abnormalities in all patients: non-visualized posterior pituitary, thin pituitary stalk, hypoplastic anterior pituitary, hypoplastic frontal and temporal lobes, thin corpus callosum and a global delay in brain myelination. All six children developed secondary microcephaly, severe global developmental delay and generalized tonic-clonic/partial seizures. Neurological examination revealed total body spastic cerebral palsy and minimal pupil response to light. All patients were dysmorphic and presented with additional features, such as gastro-oesophageal reflux and neurogenic bladder (2020. Webb EA, AlMutair A, Kelberman D, Bacchelli C, Chanudet E, Lescai F, et al. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain. 2013;136(Pt 10):3096-105.).
IGSF1 is a plasma membrane glycoprotein expressed in Rathke’s pouch and the anterior pituitary gland. Mutations in IGSF1 are X-linked and the main clinical characteristics of male patients are congenital central hypothyroidism and macroorchidism (OMIM 300888). A variable proportion of patients presented partial and transient GH, prolactin deficiency, disharmonious pubertal development and increased BMI. In one patient with GH deficiency, MRI was abnormal with hypoplasia of the corpus callosum and small stalk lesion (PSIS) (2121. Sun Y, Bak B, Schoenmakers N, van Trotsenburg AS, Oostdijk W, Voshol P, et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet. 2012;44(12):1375-81.).
CDON – GH deficiency and the link with environmental enhancers
CDON is a cell surface sonic hedgehog (SHH) binding protein that promotes SHH signaling activity. A heterozygous nonsense mutation in CDON was described in a patient with PSIS with perinatal complications: breech presentation, cesarean delivery, neonatal jaundice with increased conjugated bilirubin. The neuroimaging evaluation indicated small anterior pituitary, absent stalk, and ectopic posterior pituitary. The patients’ mother had the same mutation with a different phenotype – congenital convergent strabismus – which pointed to incomplete penetrance (2222. Bashamboo A, Bignon-Topalovic J, Rouba H, McElreavey K, Brauner R. A nonsense mutation in the hedgehog receptor CDON associated with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. 2016;101(1):12-5.). Bae and cols. reported a broad spectrum of Holoprosencephaly (HPE) phenotypes in association with CDON mutations, including hepatic cholestasis and hypotelorism (OMIM 614226) (2323. Bae GU, Domené S, Roessler E, Schachter K, Kang JS, Muenke M, et al. Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am J Hum Genet. 2011;89(2):231-40.). Incomplete penetrance has been described in many other families with pituitary anomalies with mutations involving other genes, and one mechanism proposed for this phenomenon is the gene-environment interactions (2424. Fang Q, George AS, Brinkmeier ML, Mortensen AH, Gergics P, Cheung LY, et al. Genetics of Combined Pituitary Hormone Deficiency: Roadmap into the Genome Era. Endocr Rev. 2016;37(6):636-75.). Regarding CDON, a synergy between a Cdon mutation in the mouse and ethanol leading to HPE was reported (2525. Hong M, Krauss RS. Cdon mutation and fetal ethanol exposure synergize to produce midline signaling defects and holoprosencephaly spectrum disorders in mice. PLoS Genet. 2012;8(10):e1002999.). This has also been observed with Gli2+/− and Shh+/− mice (2626. Kietzman HW, Everson JL, Sulik KK, Lipinski RJ. The teratogenic effects of prenatal ethanol exposure are exacerbated by Sonic Hedgehog or GLI2 haploinsufficiency in the mouse. PLoS One. 2014;9(2):e89448.).
Regarding the new genetic defects reported in the last ten years causing GH deficiency (Table 1), we discussed the ones with an interesting clinical picture or etiopathogenic aspect. All the genes discussed above present a strong level of evidence as disease causing.
NEW GENETIC DEFECTS ASSOCIATED WITH GH-IGFS AXIS
Although defects in GHR, IGF1 and IGF1R genes have been associated to growth disorders for a long time, the number of described individuals with heterozygous mutations in these genes has increased recently, expanding the genotype-phenotype correlations. Additionally, new genetic defects downstream of GH were described (Table 2).
Update of genotype-phenotype correlations of GHR, IGF1 and IGF1R
Autosomal-recessive mutations in the GHR gene (GHR) cause a classical picture of GH insensitivity (GHI), characterized by extreme short stature, facial dysmorphic features and IGF-1 deficiency (OMIM 262500) (2727. Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003. J Clin Endocrinol Metab. 2004;89(3):1031-44.). Several heterozygous variants in GHR have been associated with the short stature phenotype, although few have been proven as causal. Three patients with growth failure caused by heterozygous GHR mutations with a dominant-negative effect have been reported recently, totaling seven patients to date (2828. Vairamani K, Merjaneh L, Casano-Sancho P, Sanli ME, David A, Metherell LA, et al. Novel Dominant-Negative GH Receptor Mutations Expands the Spectrum of GHI and IGF-I Deficiency. J Endocr Soc. 2017;1(4):345-58.). These patients had a mild GHI phenotype which can be initially classified as idiopathic short stature (OMIM 604271). They usually presented detectable levels of IGF-1 and less severe short stature than individuals with classical GHI.
Homozygous defects in IGF1 lead to pre and postnatal short stature, microcephaly and intellectual impairment (OMIM 608747). Since the first patient described, the researchers noted that carriers of heterozygous IGF1 mutations had a reduced height and head circumference (1111. Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335(18):1363-7.). In 2010, a heterozygous frameshift mutation in IGF1 was associated with short stature and microcephaly in two siblings (2929. van Duyvenvoorde HA, van Setten PA, Walenkamp MJ, van Doorn J, Koenig J, Gauguin L, et al. Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. J Clin Endocrinol Metab. 2010;95(11):E363-7.). The mother who carried the same mutation, which had been inherited by her father, presented a milder phenotype. This fact raised the hypothesis that the probands were more affected because they were born from a heterozygous mother, with a combination of fetal and maternal IGF-1 deficiency (2929. van Duyvenvoorde HA, van Setten PA, Walenkamp MJ, van Doorn J, Koenig J, Gauguin L, et al. Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. J Clin Endocrinol Metab. 2010;95(11):E363-7.). Two years later, a heterozygous IGF1 splice site mutation was identified in a boy with postnatal short stature (height SDS of -4.0) with low IGF-1 level (SDS of -2.2). Other four relatives with short stature carried the same mutation (3030. Fuqua JS, Derr M, Rosenfeld RG, Hwa V. Identification of a novel heterozygous IGF1 splicing mutation in a large kindred with familial short stature. Horm Res Paediatr. 2012;78(1):59-66.). In 2014, a complete heterozygous deletion in IGF1 was identified in a child with short stature initially classified as idiopathic. The proband had a history of intrauterine growth restriction and was born with weight and length SDS of -1.5 and -1.2, respectively. He presented microcephaly and developmental delay (3131. Batey L, Moon JE, Yu Y, Wu B, Hirschhorn JN, Shen Y, et al. A novel deletion of IGF1 in a patient with idiopathic short stature provides insight Into IGF1 haploinsufficiency. J Clin Endocrinol Metab. 2014;99(1):E153-9.).
While defects in IGF1 are quite rare, many heterozygous defects in IGF1R gene (IGF1R) have been reported. Initially, studies described deletions encompassing IGF1R and, years later, point mutations were also identified in children with growth failure. According to a recent study, there are 36 different probably pathogenic variants in IGF1R (3232. Walenkamp MJE, Robers JML, Wit JM, Zandwijken GRJ, van Duyvenvoorde HA, Oostdijk W, et al. Phenotypic Features and Response to GH Treatment of Patients With a Molecular Defect of the IGF-1 Receptor. J Clin Endocrinol Metab. 2019;104(8):3157-71.). Classically, patients with heterozygous defects in IGF1R present pre and postnatal growth retardation, microcephaly and IGF-1 levels above the mean for age and sex (OMIM 270450). Other clinical manifestations include mental and motor development delay, cardiac defects and dysmorphic features (triangular face, clinodactyly, pectus excavatum). The degree and the prevalence of the other clinical features appear to be higher in patients with IGF1R deletions than in those carrying point mutations. In addition, among those individuals with IGF1R deletions, the severity of the clinical condition appears to be dependent on the location of the breakpoints (3232. Walenkamp MJE, Robers JML, Wit JM, Zandwijken GRJ, van Duyvenvoorde HA, Oostdijk W, et al. Phenotypic Features and Response to GH Treatment of Patients With a Molecular Defect of the IGF-1 Receptor. J Clin Endocrinol Metab. 2019;104(8):3157-71.).
Autosomal dominant defects in STAT5B
STAT5B acts as a key transcription activator of GH signaling. It has been known, since 2003, that homozygous mutations in STAT5B cause extreme short stature as part of a GH insensitivity syndrome associated with immune dysregulation (represented clinically by progressive pulmonary disease and eczema) (OMIM 245590) (3333. Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349(12):1139-47.). First-degree relatives of index cases carrying STAT5B mutations were significantly shorter than individuals from the same population (height SDS of -1.4 ± 0.8 vs. -0.4 ± 0.8; p < 0.001) (3434. Scalco RC, Hwa V, Domene HM, Jasper HG, Belgorosky A, Marino R, et al. STAT5B mutations in heterozygous state have negative impact on height: another clue in human stature heritability. Eur J Endocrinol. 2015;173(3):291-6.). Additionally, individuals who carry heterozygous mutations in STAT5B were reported to be shorter than their non-carrier family members (height SDS difference of -0.6, p = 0.009), although all of them had height within the normal range. Besides the growth impairment, eczema was observed in 4 of 32 carriers (3434. Scalco RC, Hwa V, Domene HM, Jasper HG, Belgorosky A, Marino R, et al. STAT5B mutations in heterozygous state have negative impact on height: another clue in human stature heritability. Eur J Endocrinol. 2015;173(3):291-6.). Reconstitution experiments with loss-of-function mutations inherited by recessive manner showed a lack of expression of the mutant protein (3535. Hwa V. STAT5B deficiency: impacts on human growth and immunity. Growth Horm IGF Res. 2016;28:16-20.). Therefore, the slight height reduction observed in carriers of mutations in STAT5B could be explained by partial haploinsufficiency.
Recently, three heterozygous mutations in STAT5B were reported as cause of short stature with mild GH insensitivity in three children from different families (3636. Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun. 2018;9(1):2105.). Functional evaluation revealed that those three mutations had dominant-negative effects. Mutated STAT5B proteins can be phosphorylated upon GH stimulation and can form dimers with themselves and with wild-type STAT5B proteins. In total, eleven individuals from the three families carried dominant-negative heterozygous STAT5B mutations. The degree of short stature had a high variability inter- and intra-familial and the height SDS of the probands ranged from -5.3 to -2.9. Although no one had severe immune dysregulation, the majority of affected individuals presented mild eczema (77. Amselem S, Duquesnoy P, Attree O, Novelli G, Bousnina S, Postel-Vinay MC, et al. Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med. 1989;321(15):989-95. of 1111. Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335(18):1363-7.) and elevated IgE (88. Dollé P, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M. Expression of GHF-1 protein in mouse pituitaries correlates both temporally and spatially with the onset of growth hormone gene activity. Cell. 1990;60(5):809-20. of 1111. Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335(18):1363-7.) (3636. Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun. 2018;9(1):2105.).
Gain of function mutations in STAT3
STAT3 is a cytosolic protein that acts in many physiological processes stimulated by a wide variety of cytokines and growth factors, including inteleukin-6 (IL-66. Phillips JA, Hjelle BL, Seeburg PH, Zachmann M. Molecular basis for familial isolated growth hormone deficiency. Proc Natl Acad Sci U S A. 1981;78(10):6372-5.). Studies with cell cultures systems showed that STAT3, under IL-6 activation, induces an acute-phase response in hepatoma cells and stimulates proliferation in B lymphocytes (3737. Lee CK, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17(1):63-72.). In 2014, Flanagan and cols. described that de novo germline activating STAT3 mutations were a new monogenic cause of autoimmunity (OMIM 615952). The five affected individuals had early-onset (diagnosis at <5 years) autoimmune disease (e.g. type 1 diabetes, hypothyroidism, celiac disease) and all of them had short stature (3838. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;46(8):812-4.). Short stature was also observed in 7 of 13 patients with immune dysregulation and gain-of-function (GOF) mutations in STAT3 (3939. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125(4):591-9.). In another study, a boy presented height SDS of -2.6 at 5.5 years with IGF-1 of 37 µg/L (SDS of -2.2) (4040. Sediva H, Dusatkova P, Kanderova V, Obermannova B, Kayserova J, Sramkova L, et al. Short Stature in a Boy with Multiple Early-Onset Autoimmune Conditions due to a STAT3 Activating Mutation: Could Intracellular Growth Hormone Signalling Be Compromised? Horm Res Paediatr. 2017;88(2):160-6.).
We could speculate that the growth impairment observed in those patients could be due solely to their chronic disease. However, in 2018, Gutiérrez and cols. described two unrelated children with partial GH insensitivity and immune dysregulation caused by de novo GOF STAT3 mutations (4141. Gutiérrez M, Scaglia P, Keselman A, Martucci L, Karabatas L, Domené S, et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol Cell Endocrinol. 2018;473:166-77.). One patient had height SDS of -6.4 at 2.4 years of age, with elevated serum levels of GH and prolactin associated with undetectable levels of IGF-1, while the other, at the age of 3, had height SDS of -5.4. Both presented eczema, hypothyroidism, chronic diarrhea and recurrent infections. Functional evaluation using a STAT3-responsive dual-luciferase reporter assay showed an increased reporter activity of the two STAT3 mutants in comparison to wild-type STAT3. Although mutated STAT3 were not constitutively phosphorylated, they presented delayed dephosphorylation, leading to enhanced activity. Additionally, under unstimulated conditions and under GH treatment, both GOF STAT3 variants decreased STAT5B transcriptional activity, suggesting a negative impact in the GH signaling pathway (4141. Gutiérrez M, Scaglia P, Keselman A, Martucci L, Karabatas L, Domené S, et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol Cell Endocrinol. 2018;473:166-77.).
Defects in the proteolytic cleavage of IGFBPs gene (PAPPA2)
IGF-1 can be found in the plasma bound to six IGFBPs. The ternary complex, composed by IGF-1, IGFBP-3 and ALS, is the form in which IGF-1 circulates with the highest half-life. However, to act on its receptor, IGF-1 must be released from the ternary complex (4242. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16(1):3-34.,4343. Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999;20(6):761-87.). This task is done by the metalloproteinase pregnancy-associated plasma protein A2 (PAPPA22. Waters MJ, Brooks AJ. Growth hormone receptor: structure function relationships. Horm Res Paediatr. 2011;76 Suppl 1:12-6.), a serum and tissue protease responsible for the specific proteolytic cleavage of IGFBP-3 and -5 (4444. Conover CA, Boldt HB, Bale LK, Clifton KB, Grell JA, Mader JR, et al. Pregnancy-associated plasma protein-A2 (PAPP-A2): tissue expression and biological consequences of gene knockout in mice. Endocrinology. 2011;152(7):2837-44.).
Based on the study of two different families evaluated through whole exome sequencing, mutations in PAPPA2 in an autosomal recessive inheritance were established as the cause of short stature with markedly elevated IGF-1 and IGFBP-3 concentrations (4545. Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. 2016;8(4):363-74.). One family, with two affected children, harbored a novel homozygous frameshift mutation in PAPPA2 (p.Asp643fs25*). In the other family, with three affected children, a novel missense variant (p.Ala1033Val) was observed. In vitro studies showed that both mutations led to absence of the proteolytic activity of the PAPPA2. Four of five patients had low free IGF-1 concentrations, despite the high levels of circulating IGF-1. Two children were born small for gestational age, while the others were born within the low normal range and smaller than the unaffected siblings. The growth impairment was becoming more prominent with age and other characteristics were observed, as small chins and long thin fingers (4545. Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. 2016;8(4):363-74.,4646. Argente J, Pérez-Jurado LA. Letter to the Editor: History and clinical implications of PAPP-A2 in human growth: When reflecting on idiopathic short stature leads to a specific and new diagnosis: Understanding the concept of “low IGF-I availability”. Growth Horm IGF Res. 2018;40:17-9.). Three of five affected children presented microcephaly, with no history of neuropsychomotor development delay. Bone age was consistent with chronological age in all children (4545. Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. 2016;8(4):363-74.).
Defects in the regulatory subunit of PI3K gene (PIK3R1)
The phosphatidylinositol 3 kinase (PI33. Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180(2):247-55.K) intracellular pathway is crucial to the action of IGF-1. After activation of tyrosine kinase receptor, substrates are phosphorylated and they bind to the regulatory subunit of PI3K, encoded by PIK3R1 gene (4747. Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158-66.).
In 2013, heterozygous mutations in PIK3R1 were established as the major cause of SHORT syndrome, a rare disease characterized by short stature, delayed dentition, characteristic facial features, ocular depression, lipodystrophy and hyperextensibility of joints (OMIM 269880). Several gene defects in PIK3R1 were described (nucleotides insertion or deletion, nonsense and missense variants), with prevalence of de novo inheritance. One missense variant (p.Arg649Trp), within the context of a CpG motif, was recurrent in unrelated patients (4747. Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158-66.
48. Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141-9.-4949. Chudasama KK, Winnay J, Johansson S, Claudi T, König R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013;93(1):150-7.). There was a phenotype variability among the affected patients, including the degree of the growth impairment. Some individuals had prenatal short stature, with relative microcephaly, whereas others had height in the low-normal range. The mechanism of how mutations in PIK3R1 result in the complex phenotype of SHORT syndrome is not completely understood. It is known that the regulatory subunit of PI3K has different isoforms expressed in several tissues (4747. Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158-66.
48. Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141-9.-4949. Chudasama KK, Winnay J, Johansson S, Claudi T, König R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013;93(1):150-7.).
IGF2 defects
During the prenatal period, IGF2 is expressed by the paternal allele in the placenta and has a fundamental role in promoting fetal growth. The clinical importance of the IGF-2 function was acknowledged from the study of patients with the Silver-Russell syndrome (SRS). Up to 60% of the cases of SRS are caused by the hypomethylation of the imprinted domain on chromosome 11p15.5, which leads to a decrease in the IGF2 expression. The second most frequent cause of SRS is the maternal uniparental disomy of the chromosome 7. Children with SRS classically present prenatal onset short stature, relative macrocephaly, frontal bossing, triangular face, micrognathia, clinodactily, body asymmetry and feeding problems with low body mass index (5050. Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13(2):105-24.).
In 2015, Begemann and cols. described four individuals from the same family with SRS phenotype without a recognized molecular cause. Through exome analysis, the authors identified an IGF2 nonsense mutation in all patients, inherited from their healthy fathers (OMIM 616489) (5151. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally Inherited IGF2 Mutation and Growth Restriction. N Engl J Med. 2015;373(4):349-56.). After this first report, other five studies identified variants in IGF2 gene on the paternal allele causing SRS-like (5252. Yamoto K, Saitsu H, Nakagawa N, Nakajima H, Hasegawa T, Fujisawa Y, et al. De novo IGF2 mutation on the paternal allele in a patient with Silver-Russell syndrome and ectrodactyly. Hum Mutat. 2017;38(8):953-8.
53. Liu D, Wang Y, Yang XA. De novo mutation of paternal IGF2 gene causing Silver–Russell syndrome in a sporadic patient Front Genet. 2017;8:105.
54. Abi Habib W, Brioude F, Edouard T, Bennett JT, Lienhardt-Roussie A, Tixier F, et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet Med. 2018;20(2):250-8.
55. Poulton C, Azmanov D, Atkinson V, Beilby J, Ewans L, Gration D, et al. Silver Russel syndrome in an aboriginal patient from Australia. Am J Med Genet Part A. 2018;176(12):2561-3.-5656. Rockstroh D, Pfäffle H, Le Duc D, Rößler F, Schlensog-Schuster F, Heiker JT, et al. A new p.(Ile66Serfs*93) IGF2 variant is associated with pre- and postnatal growth retardation. Eur J Endocrinol. 2019;180(1):K1-K13.). Up to now, one missense and six loss-of-function variants (33. Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180(2):247-55. frameshift, 22. Waters MJ, Brooks AJ. Growth hormone receptor: structure function relationships. Horm Res Paediatr. 2011;76 Suppl 1:12-6. nonsense and 11. Ranke MB, Wit JM. Growth hormone – past, present and future. Nat Rev Endocrinol. 2018;14(5):285-300. splice-site) in the IGF2 gene were reported. All affected children were born small for gestational age regarding weight and length, had relative macrocephaly and short stature at preschool age (height SDS ranged from -3.3 to -6.2). They had varying degrees of feeding problems and neuropsychomotor developmental delay. Five probands presented cardiac abnormalities, including patent ductus arteriosus and atrial or ventricular septal defects. Male genital abnormalities, as hypospadia, penoscrotal transposition, cryptorchidism and ambiguous genitalia were also reported (5151. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally Inherited IGF2 Mutation and Growth Restriction. N Engl J Med. 2015;373(4):349-56.
52. Yamoto K, Saitsu H, Nakagawa N, Nakajima H, Hasegawa T, Fujisawa Y, et al. De novo IGF2 mutation on the paternal allele in a patient with Silver-Russell syndrome and ectrodactyly. Hum Mutat. 2017;38(8):953-8.-5353. Liu D, Wang Y, Yang XA. De novo mutation of paternal IGF2 gene causing Silver–Russell syndrome in a sporadic patient Front Genet. 2017;8:105.,5656. Rockstroh D, Pfäffle H, Le Duc D, Rößler F, Schlensog-Schuster F, Heiker JT, et al. A new p.(Ile66Serfs*93) IGF2 variant is associated with pre- and postnatal growth retardation. Eur J Endocrinol. 2019;180(1):K1-K13.).
In addition to mutations in IGF2, Habib and cols. described new loss-of-function mutations in HMGA2 and PLAG1 genes in a cohort of suspected SRS patients. Both genes belong to the HMGA2–PLAG1–IGF2 pathway and act as positive upstream regulators of IGF2 (5454. Abi Habib W, Brioude F, Edouard T, Bennett JT, Lienhardt-Roussie A, Tixier F, et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet Med. 2018;20(2):250-8.).
All genes described above have a good level of evidence regarding their implication on GH-IGF1 axis disruption. The group of the oldest genes (GHR, IGF1 and IGF1R) in addition to IGF2 have the highest level of evidence, as there are many reports of affected individuals from different families. Heterozygous mutations in STAT5B with a dominant-negative effect and homozygous mutations in PAPPA2 also present a gene-disease clinical validity. Although defects in these genes were reported in only three and two different families, respectively, the variants segregated according to phenotype and had a convincing functional evaluation. Finally, STAT3 and PIK3R1 are also clinically relevant genes that impair the somatotropic axis. The phenotype of affected individuals are complex and there is a prevalence of the novo inheritance, reinforcing the gene-disease relationship.
CONCLUSION
Although many phenotypes associated with the GH-IGF1 axis disruption have been recognized for many decades and the most frequent genetic defects were described before 2010, the discovery of defects in new genes has increased over the last decade. This increment was mainly due to the development of large-scale parallel sequencing techniques (targeted or exome sequencing), which allowed the analysis of many genetic defects simultaneously at low cost and in a shorter time than the previous candidate gene approach. Besides the identification of new genetic defects, this innovation permitted the expansion of the genotype-phenotype correlations related to known genes, boosting the clinical use of the genetic knowledge.
Since the GH-IGF1 axis is the main regulator of growth, it is insightful that defects at any step of this pathway could lead to short stature. These genes are responsible from the GH production to IGF-1 biodisponibility and action. In this review, we focused on the most relevant new genetic defects regarding the level of evidence recommended by the Clinical Genome Resource (ClinGen) (5757. Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am J Hum Genet. 2017;100(6):895-906.).
As a future perspective, we speculate that defects in other genes which encoded intracellular signaling molecules could cause short stature. As these molecules are important to different pathways in different tissues, affected individuals would be rare and would present a multisystem phenotype. Also, we believe it will continue being a challenge to evaluate the clinical impact of variants in genes not previously associated to GH-IGF1 axis defects. This will demand careful and continuous research in this area.
REFERENCES
-
1Ranke MB, Wit JM. Growth hormone – past, present and future. Nat Rev Endocrinol. 2018;14(5):285-300.
-
2Waters MJ, Brooks AJ. Growth hormone receptor: structure function relationships. Horm Res Paediatr. 2011;76 Suppl 1:12-6.
-
3Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180(2):247-55.
-
4Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99(9):3080-92.
-
5Snell GD. Dwarf, a new Mendelian recessive character of the house mouse. Proc Natl Acad Sci U S A. 1929;15(9):733-4.
-
6Phillips JA, Hjelle BL, Seeburg PH, Zachmann M. Molecular basis for familial isolated growth hormone deficiency. Proc Natl Acad Sci U S A. 1981;78(10):6372-5.
-
7Amselem S, Duquesnoy P, Attree O, Novelli G, Bousnina S, Postel-Vinay MC, et al. Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med. 1989;321(15):989-95.
-
8Dollé P, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M. Expression of GHF-1 protein in mouse pituitaries correlates both temporally and spatially with the onset of growth hormone gene activity. Cell. 1990;60(5):809-20.
-
9Tatsumi K, Miyai K, Notomi T, Kaibe K, Amino N, Mizuno Y, et al. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet. 1992;1(1):56-8.
-
10Correa FA, Nakaguma M, Madeira JLO, Nishi MY, Abrao MG, Jorge AAL, et al. Combined pituitary hormone deficiency caused by PROP1 mutations: update 20 years post-discovery. Arch Endocrinol Metab. 2019;63(2):167-74.
-
11Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335(18):1363-7.
-
12Pinto G, Netchine I, Sobrier ML, Brunelle F, Souberbielle JC, Brauner R. Pituitary stalk interruption syndrome: a clinical-biological-genetic assessment of its pathogenesis. J Clin Endocrinol Metab. 1997;82(10):3450-4.
-
13Correa FA, Jorge AA, Nakaguma M, Canton AP, Costa SS, Funari MF, et al. Pathogenic copy number variants in patients with congenital hypopituitarism associated with complex phenotypes. Clin Endocrinol (Oxf). 2018;88(3):425-31.
-
14Zwaveling-Soonawala N, Alders M, Jongejan A, Kovacic L, Duijkers FA, Maas SM, et al. Clues for polygenic inheritance of pituitary stalk interruption syndrome from exome sequencing in 20 patients. J Clin Endocrinol Metab. 2018;103(2):415-28.
-
15Arnhold IJ, Franca MM, Carvalho LR, Mendonca BB, Jorge AA. Role of GLI2 in hypopituitarism phenotype. J Mol Endocrinol. 2015;54(3):R141-50.
-
16Franca MM, Jorge AA, Carvalho LR, Costalonga EF, Vasques GA, Leite CC, et al. Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J Clin Endocrinol Metab. 2010;95(11):E384-91.
-
17Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012;97(4):E694-9.
-
18McCabe MJ, Gaston-Massuet C, Tziaferi V, Gregory LC, Alatzoglou KS, Signore M, et al. Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab. 2011;96(10):E1709-18.
-
19Correa FA, Trarbach EB, Tusset C, Latronico AC, Montenegro LR, Carvalho LR, et al. FGFR1 and PROKR2 rare variants found in patients with combined pituitary hormone deficiencies. Endocr Connect. 2015;4(2):100-7.
-
20Webb EA, AlMutair A, Kelberman D, Bacchelli C, Chanudet E, Lescai F, et al. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain. 2013;136(Pt 10):3096-105.
-
21Sun Y, Bak B, Schoenmakers N, van Trotsenburg AS, Oostdijk W, Voshol P, et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet. 2012;44(12):1375-81.
-
22Bashamboo A, Bignon-Topalovic J, Rouba H, McElreavey K, Brauner R. A nonsense mutation in the hedgehog receptor CDON associated with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. 2016;101(1):12-5.
-
23Bae GU, Domené S, Roessler E, Schachter K, Kang JS, Muenke M, et al. Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am J Hum Genet. 2011;89(2):231-40.
-
24Fang Q, George AS, Brinkmeier ML, Mortensen AH, Gergics P, Cheung LY, et al. Genetics of Combined Pituitary Hormone Deficiency: Roadmap into the Genome Era. Endocr Rev. 2016;37(6):636-75.
-
25Hong M, Krauss RS. Cdon mutation and fetal ethanol exposure synergize to produce midline signaling defects and holoprosencephaly spectrum disorders in mice. PLoS Genet. 2012;8(10):e1002999.
-
26Kietzman HW, Everson JL, Sulik KK, Lipinski RJ. The teratogenic effects of prenatal ethanol exposure are exacerbated by Sonic Hedgehog or GLI2 haploinsufficiency in the mouse. PLoS One. 2014;9(2):e89448.
-
27Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003. J Clin Endocrinol Metab. 2004;89(3):1031-44.
-
28Vairamani K, Merjaneh L, Casano-Sancho P, Sanli ME, David A, Metherell LA, et al. Novel Dominant-Negative GH Receptor Mutations Expands the Spectrum of GHI and IGF-I Deficiency. J Endocr Soc. 2017;1(4):345-58.
-
29van Duyvenvoorde HA, van Setten PA, Walenkamp MJ, van Doorn J, Koenig J, Gauguin L, et al. Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. J Clin Endocrinol Metab. 2010;95(11):E363-7.
-
30Fuqua JS, Derr M, Rosenfeld RG, Hwa V. Identification of a novel heterozygous IGF1 splicing mutation in a large kindred with familial short stature. Horm Res Paediatr. 2012;78(1):59-66.
-
31Batey L, Moon JE, Yu Y, Wu B, Hirschhorn JN, Shen Y, et al. A novel deletion of IGF1 in a patient with idiopathic short stature provides insight Into IGF1 haploinsufficiency. J Clin Endocrinol Metab. 2014;99(1):E153-9.
-
32Walenkamp MJE, Robers JML, Wit JM, Zandwijken GRJ, van Duyvenvoorde HA, Oostdijk W, et al. Phenotypic Features and Response to GH Treatment of Patients With a Molecular Defect of the IGF-1 Receptor. J Clin Endocrinol Metab. 2019;104(8):3157-71.
-
33Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349(12):1139-47.
-
34Scalco RC, Hwa V, Domene HM, Jasper HG, Belgorosky A, Marino R, et al. STAT5B mutations in heterozygous state have negative impact on height: another clue in human stature heritability. Eur J Endocrinol. 2015;173(3):291-6.
-
35Hwa V. STAT5B deficiency: impacts on human growth and immunity. Growth Horm IGF Res. 2016;28:16-20.
-
36Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun. 2018;9(1):2105.
-
37Lee CK, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17(1):63-72.
-
38Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;46(8):812-4.
-
39Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125(4):591-9.
-
40Sediva H, Dusatkova P, Kanderova V, Obermannova B, Kayserova J, Sramkova L, et al. Short Stature in a Boy with Multiple Early-Onset Autoimmune Conditions due to a STAT3 Activating Mutation: Could Intracellular Growth Hormone Signalling Be Compromised? Horm Res Paediatr. 2017;88(2):160-6.
-
41Gutiérrez M, Scaglia P, Keselman A, Martucci L, Karabatas L, Domené S, et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol Cell Endocrinol. 2018;473:166-77.
-
42Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16(1):3-34.
-
43Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999;20(6):761-87.
-
44Conover CA, Boldt HB, Bale LK, Clifton KB, Grell JA, Mader JR, et al. Pregnancy-associated plasma protein-A2 (PAPP-A2): tissue expression and biological consequences of gene knockout in mice. Endocrinology. 2011;152(7):2837-44.
-
45Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. 2016;8(4):363-74.
-
46Argente J, Pérez-Jurado LA. Letter to the Editor: History and clinical implications of PAPP-A2 in human growth: When reflecting on idiopathic short stature leads to a specific and new diagnosis: Understanding the concept of “low IGF-I availability”. Growth Horm IGF Res. 2018;40:17-9.
-
47Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158-66.
-
48Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141-9.
-
49Chudasama KK, Winnay J, Johansson S, Claudi T, König R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013;93(1):150-7.
-
50Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13(2):105-24.
-
51Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally Inherited IGF2 Mutation and Growth Restriction. N Engl J Med. 2015;373(4):349-56.
-
52Yamoto K, Saitsu H, Nakagawa N, Nakajima H, Hasegawa T, Fujisawa Y, et al. De novo IGF2 mutation on the paternal allele in a patient with Silver-Russell syndrome and ectrodactyly. Hum Mutat. 2017;38(8):953-8.
-
53Liu D, Wang Y, Yang XA. De novo mutation of paternal IGF2 gene causing Silver–Russell syndrome in a sporadic patient Front Genet. 2017;8:105.
-
54Abi Habib W, Brioude F, Edouard T, Bennett JT, Lienhardt-Roussie A, Tixier F, et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet Med. 2018;20(2):250-8.
-
55Poulton C, Azmanov D, Atkinson V, Beilby J, Ewans L, Gration D, et al. Silver Russel syndrome in an aboriginal patient from Australia. Am J Med Genet Part A. 2018;176(12):2561-3.
-
56Rockstroh D, Pfäffle H, Le Duc D, Rößler F, Schlensog-Schuster F, Heiker JT, et al. A new p.(Ile66Serfs*93) IGF2 variant is associated with pre- and postnatal growth retardation. Eur J Endocrinol. 2019;180(1):K1-K13.
-
57Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am J Hum Genet. 2017;100(6):895-906.
-
58Takagi M, Nagasaki K, Fujiwara I, Ishii T, Amano N, Asakura Y, et al. Heterozygous defects in PAX6 gene and congenital hypopituitarism. Eur J Endocrinol. 2015;172(1):37-45.
-
59Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, et al. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat. 2010;31(10): 1142-54.
-
60Karaca E, Buyukkaya R, Pehlivan D, Charng WL, Yaykasli KO, Bayram Y, et al. Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. 2015;100(1):E140-7.
-
61Tatsi C, Sertedaki A, Voutetakis A, Valavani E, Magiakou MA, Kanaka-Gantenbein C, et al. Pituitary stalk interruption syndrome and isolated pituitary hypoplasia may be caused by mutations in holoprosencephaly-related genes. J Clin Endocrinol Metab. 2013;98(4):E779-84.
-
62Gaston-Massuet C, McCabe MJ, Scagliotti V, Young RM, Carreno G, Gregory LC, et al. Transcription factor 7-like 1 is involved in hypothalamo-pituitary axis development in mice and humans. Proc Natl Acad Sci U S A. 2016;113(5):E548-57.
-
Grants: this work was supported by Grants 2013/03236-5 (to A.A.L.J.) from the São Paulo Research Foundation (Fapesp), Grant 304678/2012-0 (to A.A.L.J.) from the National Council for Scientific and Technological Development (CNPq) and by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (Capes) – Finance Code 1658266 (to GAV).
Publication Dates
-
Publication in this collection
10 Jan 2020 -
Date of issue
Nov-Dec 2019
History
-
Received
3 Sept 2019 -
Accepted
19 Nov 2019