ABSTRACT
Considering that thiol-containing enzymes like kinases are critical for several metabolic pathways and energy homeostasis, we investigated the effects of cystine dimethyl ester and/or cysteamine administration on kinases crucial for energy metabolism in the kidney of Wistar rats. Animals were injected twice a day with 1.6 µmol/g body weight cystine dimethyl ester and/or 0.26 µmol/g body weight cysteamine from the 16th to the 20th postpartum day and euthanized after 12 hours. Pyruvate kinase, adenylate kinase, creatine kinase activities and thiol/disulfide ratio were determined. Cystine dimethyl ester administration reduced thiol/disulfide ratio and inhibited the kinases activities. Cysteamine administration increased the thiol/disulfide ratio and co-administration with cystine dimethyl ester prevented the inhibition of the enzymes. Regression between the thiol/disulfide ratio, and the kinases activities were significant. These results suggest that redox status may regulate energy metabolism in the rat kidney. If thiol-containing enzymes inhibition and oxidative stress occur in patients with cystinosis, it is possible that lysosomal cystine depletion may not be the only beneficial effect of cysteamine administration, but also its antioxidant and thiol-protector effect.
Key words:
cysteamine; cystine dimethyl ester; kinases activities; renal energy metabolism
INTRODUCTION
A variety of metabolic processes has been shown to be altered significantly under conditions in which the thiol/disulfide ratio changes. The activity of some enzymes can be modified by the reversible oxidation of catalytically important protein thiols to disulfides. Enzyme thiols may be oxidized in vivo by reaction with cellular disulfide, like oxidized glutathione, cystine, cystamine or oxidized thioredoxin. This oxidation could increase, decrease, or have no effect on enzyme activity (Moriarty-Craige and Jones 2004JONES DP, GO YM, ANDERSON CL, ZIEGLER TR, KINKADE JR JM AND KIRLIN WG. 2004. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J 18: 1246-1248.).
Pyruvate kinase (PK; EC 2.7.1.40), creatine kinase (CK; EC 2.7.3.2) and adenylate kinase (AK; EC 2.7.4.3) are thiol-containing enzymes critical for energy metabolism in several mammalian tissues. PK is a crucial enzyme of glucose metabolism, the main pathway that provides energy for kidney function (Hall and Cottam 1978HALL ER AND COTTAM GL. 1978. Isoenzymes of pyruvate kinase in vertebrates: their physical, chemical, kinetic and immunological properties. Int J Biochem 9: 785-793.). CK is a thiol-containing enzyme that catalyzes the reversible transfer of the phosphoryl group from phosphocreatine to ADP, regenerating ATP. There are distinct CK isoenzymes, which are compartmentalized specifically in the places where energy is liberated (mitochondria) or utilized (cytosol) (Wallimann et al. 1992WALLIMANN T, WYSS M, BRDICZKA D, NICOLAY K AND EPPENBERGER HM. 1992. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J 281: 21-40.). AK catalyzes the reversible transfer of phosphoryl between ATP, ADP, and AMP (Prices et al. 1975PRICES NC, COHN M AND SCHIRMER RH. 1975. Fluorescent and Spin Label Probes of the Environments of the Sulfhydryl Groups of Porcine Muscle Adenylate Kinase. J Biol Chem 250: 644-652.). AK, PK, and CK are critical for the enzymatic phosphoryl transfer network, in other words, responsible for the transfer of the γ-phosphoryl of ATP from mitochondria to the cytosol (Dzeja and Terzic 2003DZEJA PP AND TERZIC A. 2003. Phosphotransfer networks and cellular energetics. J Exp Biol 206: 2039-2047.).
Cystine (CySS) is the oxidized dimer of cysteine and is an oxidant substance. Its liberation causes a diminution of ATP levels and the ratio of reduced glutathione/oxidized glutathione (GSH/GSSG) and increased susceptibility to undergo apoptosis (Ben-Nun et al. 1993, Foreman et al. 1995FOREMAN JW, BENSON LL, WELLONS M, AVNER ED, SWEENEY W, NISSIM L AND NISSIM I. 1995. Metabolic studies of rat renal tubule cells loaded with cystine: the cystine dimethyl ester model of cystinosis. J Am Soc Nephrol 6: 269-272., Coor et al. 1991COOR C, SALMON RF, QUIGLEY R, MARVER D AND BAUM M. 1991. Role of adenosine- triphosphate (ATP) and Na+, K+ -ATPase in the inhibition of proximal tubule transport with intracellular cystine loading. J Clin Invest 87: 955-961., Park and Thoene 2005PARK MA AND THOENE JG. 2005. Potential role of apoptosis in development of the cystinotic phenotype. Pediatr Nephrol 20: 441-446.). Cysteamine (CSH) has a free thiol group and is known as a reducing agent (Cappel and Gilbert 1986CAPPEL RE AND GILBERT HF. 1986. Cooperative behavior in the thiol oxidation of rabbit muscle glycogen phosphorylase in cysteamine/cystamine redox buffers. J Biol Chem 261: 15378-15384.) and efficiently decreases apoptotic cell death and cell oxidation in vitro (Emma et al. 2014EMMA F ET AL. 2014. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant 29: iv87-iv94.).
We have already demonstrated that CySS or cystine dimethyl ester inhibit thiol-containing kinases in the porcine retina, and in the brain of rats, and CSH prevents in vivo and in vitro these effects (Rech et al. 2006RECH VC, ATHAYDES GA, FEKSA LR, DORNELLES PK, RODRIGUES-JUNIOR V, DUTRA-FILHO CS, DE SOUZA WYSE AT, WAJNER M AND WANNMACHER CM. 2006. Inhibition of creatine kinase activity by cystine in the kidney of young rats. Pediatr Res 1124: 190-195., Pereira Oliveira et al. 2007, Rech et al. 2008). Considering that administration of CDME and/or CSH may alter thiol/disulfide status, the main objective of the present study was to investigate the in vivo effects of thiol/disulfide ratio variation on CK, PK and AK activities in the kidney of young Wistar rats.
MATERIALS AND METHODS
ANIMALS AND REAGENTS
Wistar rats bred in the Department of Biochemistry, UFRGS, were used in the experiments. The animals were kept with dams until they were euthanized. The dams had free access to water and to a standard commercial chow (Supra, Porto Alegre, RS, Brazil) containing 20.5 protein (predominantly soybean supplemented with methionine), 54% carbohydrate, 4.5% fiber, 4% lipids, 7% ash and 10% moisture. Temperature was maintained at 24 ± 1 °C, with a 12-12 h light-dark cycle. The “Principles of Laboratory Animal Care” (NIH publication no. 80-23, revised 1996; http://www.nap.edu/readingroom/books/labrats/) were followed in all the experiments, and the Ethics Committee for Animal Research of the Universidade Federal do Rio Grande do Sul approved the experimental protocol. All efforts were made to minimize animal suffering and to use only the number of animals necessary to produce reliable scientific data. All chemicals were purchased from Sigma Chemical Co., St Louis, MO, USA.
TREATMENT OF THE ANIMALS
Twenty-eight rats were randomly separated into four groups (n=7) and were treated from the 16th to the 20th postpartum day; at this age, Wistar rat development is equivalent to the development of a 6-year-old child (Sengupta 2013SENGUPTA P. 2013. The Laboratory Rat: Relating Its Age With Human’s. Int J Prev Med 4: 624-630.). The animals were kept with dams while receiving administration of 20 µL/g body weight of one of the following buffered solutions (pH 7.4), twice a day at 12 hours interval: 0.85 g% saline (Control group), 80 mM CDME (CDME group), 13 mM CSH (CSH group), or 80 mM CDME + 13 mM CSH (CDME + CSH group). CSH was administered subcutaneously and saline or CDME were administered intraperitoneally. CDME dose was equivalent to that used to load lysosomes in adult rats (Ben-Nun et al. 1993); CSH dose was equivalent to that used for the treatment of the patients (Gahl 2003GAHL WA. 2003. Early oral cysteamine therapy for nephropathic cystinosis. Eur J Pediatr 162: 38-41.). Rats were euthanized in the 21st day, twelve hours after the last injection, and the kidneys removed for thiols, disulfides, AK, CK and PK determination.
PREPARATION OF KIDNEY TISSUE
Treated animals were euthanized by decapitation without anesthesia, the kidneys were rapidly removed and decapsulated. Time elapsed between decapitation and kidney decapsulation was less than 1 min. The kidney was homogenized with a Teflon-glass homogenizer (B. Braun Biotech International, Melsungen, Germany) in 5 volumes of ice-cold SET buffer (0.32 M sucrose, 1 mM EGTA, 10 mM Tris-HCl), pH 7.4. The homogenate was centrifuged at 800 x g for 10 min in a Sorvall RC5B Plus centrifuge (Thermo Fisher Scientific, Pittsburgh, Kansas, USA) and the pellet was discarded. An aliquot of the supernatant was separated for AK activity determination and the rest was centrifuged at 10,000 x g for 15 min. The supernatant solution, containing cytosol and other cellular components such as endoplasmic reticulum and lysosomes, was collected for determination of PK, cytosolic CK activities and thiol and disulfide content. The pellet, enriched in mitochondria, was washed twice with the same Tris-sucrose isotonic buffer, resuspended in 100 mM Tris-HCl buffer, pH 7.5, containing 15 mM MgSO4 for determination of mitochondrial CK activity and thiol and disulfide content. All steps were performed at 4 ºC. The subcellular fractions were stored for no more than 1 week at -70 ºC when the assay was not carried out immediately. The mitochondrial fraction was frozen and thawed three times immediately before the assay, to break mitochondrial membranes, facilitating the interaction between CK and the substrates.
CREATINE KINASE ACTIVITY ASSAY
The reaction mixture contained the following final concentrations: 60 mM Tris-HCl buffer, pH 7.5, 7 mM phosphocreatine, 9 mM MgSO4, and approximately 1 µg protein in a final volume of 0.1 mL. After a pre-incubation by 5 min at 37 ºC, the enzymatic reaction was started by the addition of 0.42 µmol of ADP. The reaction was stopped after the incubation for 10 minutes by the addition of 1 µmol p-hydroxymercuribenzoic acid. The reagent concentrations and the incubation time were chosen to assure linearity of the enzymatic reaction. Appropriate controls were carried out to discount chemical hydrolysis of phosphocreatine and the amount of creatine already present in the enzymatic material. The creatine formed by the enzymatic action was estimated according to the colorimetric method of Hughes (1962HUGHES BP. 1962. A method for the estimation of serum creatine kinase and its use in comparing creatine kinase and aldolase activity in normal and pathological sera. Clin Chim Acta 7: 597-603.). The color was developed by the addition of 0.1 mL 2% α-naphtol and 0.1 mL 0.05% diacetyl in a final volume of 1 mL and read after 20 minutes at 540 nm. Results were expressed as nmol of creatine formed per min per mg protein.
PYRUVATE KINASE ACTIVITY ASSAY
PK activity was assayed essentially as described by Leong et al. (1981LEONG SF, LAI JC, LIM L AND CLARK JB. 1981. Energy-metabolising enzymes in brain regions of adult and aging rats. J Neurochem 37: 1548-1556.). The incubation medium consisted of 0.1 M Tris-HCl buffer, pH 7.5, 10 mM MgCl2, 0.16 mM NADH, 75 mM KCl, 5.0 mM ADP, 7 units of lactate dehydrogenase (LDH), 0.1% (v/v) Triton X-100, and 10 µL of the mitochondria-free supernatant in a final volume of 0.5 mL. The reaction was started after 30 min of pre-incubation at 37 ºC by the addition of 1.0 mM phosphoenolpyruvate. NADH oxidation was recorded spectrophotometrically during 2 min at 340 nm. All assays were performed in triplicate at 25 ºC. Reagents concentration and assay time (2 min) were chosen to assure the linearity of the reaction. CDME or CSH did not interfere with LDH activity or with spectrophotometric readings. Results were expressed as nmol of pyruvate formed per min per mg of protein.
ADENYLATE KINASE ACTIVITY ASSAY
AK activity was measured with a coupled enzyme assay with hexokinase (HK) and glucose 6-phosphate dehydrogenase (G6PD), according to Dzeja et al. (1999DZEJA PP, VITKEVICIUS KT, REDFIELD MM, BURNETT JC AND TERZIC A. 1999. Adenylate Kinase-Catalyzed Phosphotransfer in the Myocardium: Increased Contribution in Heart Failure. Circ Res 84: 1137-1143.). The reaction mixture consisted of 100 mM KCl, 20 mM HEPES, 20 mM glucose, 4 mM MgCl2, 2 mM NADP+, 1 mM EDTA, 4.5 U/ml HK, 2 U/mL of G6PD and 1µg of protein homogenate. The reaction was initiated by the addition of 2 mM ADP and the reduction of NADP+ was followed at 340 nm in a spectrophotometer for 3 minutes. ADP, NADP+, HK and G6PD were dissolved in water. Reagents concentration and assay time (3 min) were chosen to assure the linearity of the reaction. Results were expressed in nmol of ATP formed per min per mg of protein.
DETERMINATION OF THIOLS AND DISULFIDES
Thiols and disulfides were determined essentially according to Zahler and Cleland (1968ZAHLER WL AND CLELAND WW. 1968. A specific and sensitive assay for disulfides. J Biol Chem 243: 716-719.). The reaction medium consisted of 0.1 mL of 50 mM Tris buffer pH 9.0, 0.1 mL of 3 mM DTT (dithiothreitol), and 0.2 mL of cytosolic or mitochondrial fraction. After 20 min at room temperature, 0.2 mL of 1.0 M Tris buffer pH 8.1 and 1.5 mL of sodium arsenite were added. After 2 min, 0.1 mL of 3 mM DTNB (5,5’-dithiobis -2-nitrobenzoic acid) in 50 mM acetate buffer pH 5.0 were added and the absorbance at 412 nm was recorded for 3 min. For thiol determination, the same procedure was performed, omitting DTT and sodium arsenite. The disulfide content was calculated through the difference between the two determinations. A mixture of cysteine and cystine was used as the standard. Results were expressed as mmol of cystine (disulfide) or mmol of cysteine (thiol) per mg of protein.
PROTEIN DETERMINATION
Protein content was determined by the method of Lowry et al. (1951LOWRY OH, ROSEBROUGH NJ AND FARR AL. 1951. Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275.) using bovine serum albumin as the standard.
STATISTICAL ANALYSIS
Data from the weight of the animals were analyzed by repeated measures ANOVA followed by the Tukey test when the F values were significant. Data from kidney weight, protein content in the kidney, enzyme activities, and thiol/disulfide ratios, were analyzed by two-way ANOVA (presence or absence of CDME; presence or absence of CSH). Post hoc analysis of a significant interaction, when required, was performed by the Tukey test. The effect of thiol/disulfide ratio on the enzymes activities were analyzed by linear regression. All data were analyzed by the Statistical Package for the Social Sciences software (SPSS 12.0 for Windows) (Leech et al. 2005LEECH NL, BARRETT KC AND MORGAN GA. 2005. SPSS for intermediate statistics. Use and interpretation, 2nd ed., London, England: Lawrence Erlbaum Associates Publishers.).
RESULTS
The animals were treated with CDME and /or CSH for six days. Treatment did not affect body weight, kidney weight or protein content of kidney. Body weight (g) was 32±2 (controls); 30±2 (CSH); 30±2 (CDME); 32±1 (CDME + CSH). Kidney weight (mg) was 452±92 (Control); 440±25 (CDME); 436±25 (CSH) and 460±63 (CDME + CSH). Protein content (mg per g of wet weight) was 34±5 (Control); 36±4 (CDME); 34±5 (CSH) and 37±6 (CDME + CSH). Two-ways ANOVA showed no interaction between CDME and CSH in body weight [F (1, 25) = 1.62; p > 0.05], kidney weight [F (1, 25) = 1.26; p > 0.27], as well as protein content [F (1, 25) = 0.57; p > 0.45] at the end of the treatment, indicating that the biochemical differences observed between the groups cannot be attributed to protein loss.
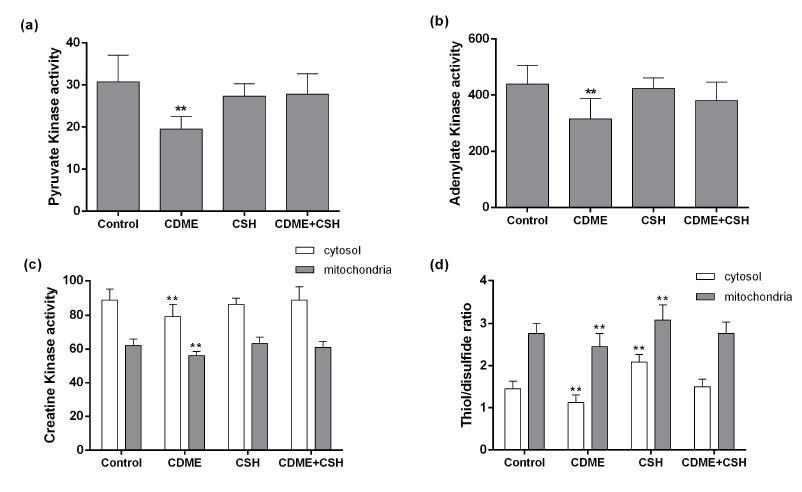
Two-ways ANOVA for CDME by CSH interaction showed that co-administration of CSH prevented the effect of CDME loading on PK activity [F (1, 25) = 4.49; p < 0.05], AK activity [F(1, 25) = 4.42; p < 0.05], mitochondrial CK activity [F (1, 25) = 21.47; p < 0.001], and cytosolic CK activity [F (1, 25) = 5.12; p < 0.05]. Post hoc comparison between means indicated that CDME administration reduced the activities of PK (Figure 1a), AK (Figure 1b) and subcellular fractions of CK (Figure 1c), whereas CSH co-administration prevented the diminution of the enzymes activities. CDME by CSH interaction was significant for cytosolic [F (1, 25) = 3.77; p < 0.05], and mitochondrial [F (1, 25) = 4.29; p < 0.05] thiol/disulfide ratio. Post hoc comparison between means indicated that CDME administration reduced thiol/disulfide ratio in the two subcellular fractions and CSH co-administration increased this ratio (Figure 1d).
Effect of CDME and / or CSH administration on (a) PK activity of kidney from young rats. PK activity is expressed as nmol of pyruvate formed per min per mg of protein. (b) AK activity of kidney from young rats. AK activity is expressed as nmol of ATP formed per min per mg of protein. (c) CK activity of kidney from young rats. CK activity is expressed as nmol of creatine formed per min per mg of protein. (d) Thiol/disulfide ratio of kidney from young rats. Data are mean ± SD (n=7 animals per group). **p < 0.01 compared to the other groups (Tukey test).
Linear regression between thiol/disulfide ratio and the enzymes activities were significant for all kinases studied, strongly suggesting that the kinases activities may be regulated by the oxidative status of the subcellular ambient where the enzymes acts: PK: [F (1, 27) = 15.51; β = 0.66; p < 0.01] (Figure 2a); AK: [F (1, 27) = 6.63; β = 0.45; p < 0.01] (Figure 2b); mitochondrial CK: [F (1, 27) = 24.62; β = 0.71; p < 0.01] (Figure 2c); cytosolic CK: [F (1, 27) = 8.49; β = 0.51; p < 0.01] (Figure 2d).
Linear regression between SH/SS ratio (a) in the cytosolic fraction and PK activity. (b) in the hole homogenate (hom) and AK activity. (c) in the mitochondrial fraction and CK mit activity. (d) in the cytosolic fraction and CK cyt activity. (n=7 animals per group).
DISCUSSION
In the present work, we investigated the effects of the administration of CDME, CSH, or CDME + CSH on PK, AK, and CK activities of the kidney from young rats. We observed that CDME administration reduced the activities of the thiol-containing enzymes, and co-administration of CDME plus CSH prevented the enzyme activities diminution. We also observed that CDME administration reduced thiol/disulfide ratio and that co-administration of CSH prevented the reduction of this ratio. Linear regression between thiol/disulfide ratio and the enzymes activity in the same subcellular fraction was significant for the kinases. These results suggest that thiol/disulfide status may be an important mechanism regulating kinases activity and consequently energy homeostasis in the kidney. Methanol released from CDME hydrolysis in the cells has probably no significant toxic effect because other amino acid esters are not harmful to the cells (Salmon and Baum 1990SALMON RF AND BAUM M. 1990. Intracellular cystine loading inhibits transport in the rabbit proximal convoluted tubule. J Clin Invest 85: 340-344.).
During protein synthesis, its folding in the cell is accomplished by enzymes that catalyze thiol/disulfide exchange reactions, including disulfide formation, reduction, and isomerization and by chaperones that prevent aggregation (Wilkinson and Gilbert 2004, Hudson et al. 2014HUDSON DA, GANNON SA AND THORPE C. 2014. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radical Biol Med 80: 171-182.). Protein disulfide isomerases and chaperons are found in the eukaryotic endoplasmic reticulum ensuring that disulfides connect the proper cysteines and that the folding protein does not make wrong interactions. Misfolding of a protein can alter its function, and accumulated aggregates can also interfere with cell function. The eukaryotic cell prevents protein misfolding maintaining the redox environment (Hwang et al. 1992HWANG C, SINSKEY AJ AND LODISH HF. 1992. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257: 1496-1502.) and using the endoplasmic reticulum (ER) with enzymes of disulfide formation and isomerization (Wilkinson and Gilbert 2004).
Released proteins from ER need maintain their correct fold through maintenance of an adequate thiol/disulfide status. Chicken liver fatty acid synthase is rapidly inactivated and cross-linked by incubation with low concentrations of common biological disulfides, and the inactive enzyme is promptly and completely reactivated, and the cross-linking is completely reversed, by incubation of the enzyme with thiols (Walters and Gilbert 1986WALTERS DW AND GILBERT HF. 1986. Thiol/disulfide redox equilibrium and kinetic behavior of chicken liver fatty acid synthase. J Biol Chem 261: 13135-13143.).
The redox state of the secretory pathway is more oxidative than that of the cytosol. The ratio of reduced glutathione to the disulfide form (GSH/GSSG) within the secretory pathway range from 1:1 to 3:1, whereas the overall cellular GSH/GSSG ratio range from 30:1 to 100:1. The preferential transport of GSSG compared to GSH into the ER lumen may contribute to this redox compartmentation (Hwang et al. 1992HWANG C, SINSKEY AJ AND LODISH HF. 1992. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257: 1496-1502.).
The cysteine/cystine represents one of the major cell thiol/disulfide systems and is involved in the regulation of several metabolic pathways and the cellular redox state. The decrease of cystinosin activity causes accumulation of cystine in lysosomes, causing cystinosis, a lethal disease affecting mainly the kidneys (Bellomo et al. 2010BELLOMO F, CORALLINI S, PASTORE A, PALMA A, LAURENZI C, EMMA F AND TARANTA A. 2010. Modulation of CTNS gene expression by intracellular thiols. Free Radical Biol Med 48: 865-872.). Data obtained using in vitro models of cystinosis show to abnormal cysteinylation of proapoptotic kinases, altered cell redox state and decreased the reduced glutathione synthesis, which can impair mitochondrial activity, although mitochondria do not seem to be compromised in cystinosis (Laube et al. 2006LAUBE GF, SHAH V, STEWART VC, HARGREAVES IP, HAQ MR, HEALES SJR AND VAN’T HOFF WG. 2006. Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr Nephrol 21: 503-509., Levtchenko et al. 2006LEVTCHENKO EN, WILMER MJ, JANSSEN AJ, KOENDERINK JB, VISCH HJ, WILLEMS PH, DE GRAAF-HESS A, BLOM HJ, VAN DEN HEUVEL HP AND MONNENS LA. 2006. Decreased intracellular, ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res 59: 287-292., Park et al. 2006PARK MA, PEJOVIC V, KERISIT KG, JUNIUS S AND THOENE JG. 2006. Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase C delta. J Am Soc Nephrol 17: 3167-3175.). Although CDME load was used as a model of cystinosis, the elicited effects are considered toxic, and this load is no longer used as a reliable model of cystinosis (Wilmer et al. 2007WILMER MJ, WILLEMS PH, VERKAART S, VISCH HJ, DE GRAAF-HESS A, BLOM HJ, MONNENS LA, VAN DEN HEUVEL LP AND LEVTCHENKO EN. 2007. Cystine dimethylester model of cystinosis: still reliable? Pediatr Res 62: 151-155.). On the other hand, in human cystinosis proximal tubular epithelial cells no overall decrease in glycolytic activity could be demonstrated (Wilmer et al. 2011).
Assessment of the major cellular thiol/disulfide systems in different cellular compartments indicated that individual signaling and control events occur through discrete redox pathways (Jones et al. 2004JONES DP, GO YM, ANDERSON CL, ZIEGLER TR, KINKADE JR JM AND KIRLIN WG. 2004. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J 18: 1246-1248., 2006). A complex interaction exists between extracellular redox control and cellular redox control, with extracellular redox ultimately dependent upon the cellular redox state. Two principal systems maintain cellular thiol/disulfide redox state: GSH and thioredoxin. These systems are complementary but also have overlapping activities that provide a partial redundancy in their functions. GSH is well suited for functions in detoxifications, inter-organ cysteine homeostasis, and redox control. Thioredoxin is ideally suited for reduction of protein disulfides, sulfoxides, and sulfenic acids, but is also capable of peroxide elimination and is used for deoxyribonucleotide biosynthesis (Moriarty-Craige and Jones 2004). On the other hand, the diversity of beneficial effects of lipoic acid in a variety of tissues can be envisaged regarding thiol/disulfide exchange reactions that modulate the environment’s redox and energy status. Thiol/disulfide exchange reactions dependent on lipoic acid appear critical for the modulation of proteins involved in cell signaling and transcription factors (Packer and Cadenas 2011PACKER L AND CADENAS E. 2011. Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J Clin Biochem Nutr 48: 26-32.).
A spatially arranged intracellular enzymatic network, catalyzed by CK, AK, and glycolytic enzymes, in especial PK, supports high-energy phosphoryl transfer and signal communication between ATP-generating and ATP-consuming/ATP-sensing processes (Wallimann et al. 1992WALLIMANN T, WYSS M, BRDICZKA D, NICOLAY K AND EPPENBERGER HM. 1992. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J 281: 21-40., Dzeja et al. 1999DZEJA PP, VITKEVICIUS KT, REDFIELD MM, BURNETT JC AND TERZIC A. 1999. Adenylate Kinase-Catalyzed Phosphotransfer in the Myocardium: Increased Contribution in Heart Failure. Circ Res 84: 1137-1143.). These dynamic metabolic networks contribute to efficient intracellular energetic communication, keeping the balance between cellular ATP consumption and production, maintaining a strong, energetic homeostasis (Dzeja and Terzic 2009, Chung et al. 2010CHUNG S, ARRELL DK, FAUSTINO RS, TERZIC A AND DZEJA PP. 2010. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol 48: 725-734.). Phosphoryl flux through the integrated phosphoryl transfer systems tightly correlates with cellular functions, indicating a critical role of such networks in an efficient energy transfer and distribution, thereby maintaining the cellular energy homeostasis (Dzeja and Terzic 2003).
The decrease of PK activity observed in the present work could reduce pyruvate levels inside the renal cells, diminishing ATP production. Besides, pyruvate is an effective anti-inflammatory and anti-oxidant endogenous molecule (Das 2006DAS UN. 2006. Pyruvate is an endogenous anti-inflammatory and anti-oxidant molecule. Med Sci Monit 12: 79-84.), suggesting that pyruvate reduction may induce cell death (Berry and Toms 2006BERRY EV AND TOMS NJ. 2006. Pyruvate and oxaloacetate limit zinc-induced oxidative HT-22 neuronal cell injury. Neurotoxicology 27: 1043-1051.). Changes in glucose metabolism closely correlate with pathogenesis progression and are intimately linked to redox signaling (Dodson et al. 2013DODSON M, DARLEY-USMAR V AND ZHANG J. 2013. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med 63: 207-221.).
Creatine is important as antioxidant and as the most important endogenous substance for phosphoryl transference from mitochondria to the cytosol (Wallimann et al. 1992WALLIMANN T, WYSS M, BRDICZKA D, NICOLAY K AND EPPENBERGER HM. 1992. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J 281: 21-40.). CK inhibition diminishes the velocity of phosphoryl transference, reducing instantaneous creatine pool in cytosol and mitochondria, causing an alteration in the energy homeostasis (Nasrallah et al. 2010NASRALLAH F, FEKI M AND KAABACHI N. 2010. Creatine and creatine deficiency syndromes: biochemical and clinical aspects. Pediatr Nephrol 42: 163-171.). Both enzymes, CK and AK, are intimately associated in such a way that when one enzyme activity is reduced, the activity of the other enzyme is enhanced (Dzeja et al. 2002DZEJA PP, BORTOLON R, PEREZ-TERZIC C, HOLMUHAMEDOV EL AND TERZIC A. 2002. Energetic communication between mitochondria and nucleus directed by catalyzed phosphotransfer. Proc Natl Acad Sci USA 99: 10156-10161.). Diminution of cytosolic CK and AK activities reduces energy homeostasis, partially compensated through glycolytic phosphoryl transfer (Janssen et al. 2003JANSSEN E, TERZIC A, WIERINGA B AND DZEJA PP. 2003. Impaired intracellular energetic communication in muscles from creatine kinase and adenylate kinase (M-CK/AK1) double knock-out mice. J Biol Chem 278: 30441-30449.). Therefore, the simultaneous inhibition of CK, AK, and PK could disrupt energy homeostasis with severe consequences for cell function and survival. The diminished activity of PK, AK, and CK, and possibly of others thiol-containing enzymes, might at least partially explain the decreased ATP content found in cells loaded with CDME (Coor et al. 1991COOR C, SALMON RF, QUIGLEY R, MARVER D AND BAUM M. 1991. Role of adenosine- triphosphate (ATP) and Na+, K+ -ATPase in the inhibition of proximal tubule transport with intracellular cystine loading. J Clin Invest 87: 955-961.).
In the present work, we are demonstrating that CDME loading reduces PK, AK and CK activities in the kidney of young rats and CSH co-administration prevents this reduction. Linear regression analyses suggest that thiol/disulfide ratio might be responsible for the enzymes activity variation. Considering the limitations of CDME model of cystinosis, further studies are necessary to evaluate the status of energy homeostasis in patients affected by cystinosis before and after CSH treatment. If thiol-containing enzymes inhibition and oxidative stress occur in patients with cystinosis, it is possible that lysosomal cystine depletion may be not the only beneficial effect of CSH administration, but also its antioxidant and thiol-protector effect.
ACKNOWLEDGMENTS
This work was supported in part by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-Brazil), Fundação de Amparo à Pesquisa do Rio Grande do Sul (FAPERGS, RS-Brazil) and Programa de Núcleos de Excelência (PRONEX-CNPq /FAPERGS -Brazil).
REFERENCES
- BELLOMO F, CORALLINI S, PASTORE A, PALMA A, LAURENZI C, EMMA F AND TARANTA A. 2010. Modulation of CTNS gene expression by intracellular thiols. Free Radical Biol Med 48: 865-872.
- BEN-NUN A, BASHAN N, POTASHNIK R, COHEN-LURIA R AND MORAN A. 1993. Cystine loading induces Fanconi’s syndrome in rats: in vivo and vesicle studies. Am J Physiol 265: 839-844.
- BERRY EV AND TOMS NJ. 2006. Pyruvate and oxaloacetate limit zinc-induced oxidative HT-22 neuronal cell injury. Neurotoxicology 27: 1043-1051.
- CAPPEL RE AND GILBERT HF. 1986. Cooperative behavior in the thiol oxidation of rabbit muscle glycogen phosphorylase in cysteamine/cystamine redox buffers. J Biol Chem 261: 15378-15384.
- CHUNG S, ARRELL DK, FAUSTINO RS, TERZIC A AND DZEJA PP. 2010. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol 48: 725-734.
- COOR C, SALMON RF, QUIGLEY R, MARVER D AND BAUM M. 1991. Role of adenosine- triphosphate (ATP) and Na+, K+ -ATPase in the inhibition of proximal tubule transport with intracellular cystine loading. J Clin Invest 87: 955-961.
- DAS UN. 2006. Pyruvate is an endogenous anti-inflammatory and anti-oxidant molecule. Med Sci Monit 12: 79-84.
- DODSON M, DARLEY-USMAR V AND ZHANG J. 2013. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med 63: 207-221.
- DZEJA PP, BORTOLON R, PEREZ-TERZIC C, HOLMUHAMEDOV EL AND TERZIC A. 2002. Energetic communication between mitochondria and nucleus directed by catalyzed phosphotransfer. Proc Natl Acad Sci USA 99: 10156-10161.
- DZEJA PP AND TERZIC A. 2003. Phosphotransfer networks and cellular energetics. J Exp Biol 206: 2039-2047.
- DZEJA PP AND TERZIC A. 2009. Adenylate kinase and AMP signaling networks: metabolic monitoring, signal communication and body energy sensing. Int J Mol Sci 10: 1729-1772.
- DZEJA PP, VITKEVICIUS KT, REDFIELD MM, BURNETT JC AND TERZIC A. 1999. Adenylate Kinase-Catalyzed Phosphotransfer in the Myocardium: Increased Contribution in Heart Failure. Circ Res 84: 1137-1143.
- EMMA F ET AL. 2014. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant 29: iv87-iv94.
- FOREMAN JW, BENSON LL, WELLONS M, AVNER ED, SWEENEY W, NISSIM L AND NISSIM I. 1995. Metabolic studies of rat renal tubule cells loaded with cystine: the cystine dimethyl ester model of cystinosis. J Am Soc Nephrol 6: 269-272.
- GAHL WA. 2003. Early oral cysteamine therapy for nephropathic cystinosis. Eur J Pediatr 162: 38-41.
- HALL ER AND COTTAM GL. 1978. Isoenzymes of pyruvate kinase in vertebrates: their physical, chemical, kinetic and immunological properties. Int J Biochem 9: 785-793.
- HUDSON DA, GANNON SA AND THORPE C. 2014. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radical Biol Med 80: 171-182.
- HUGHES BP. 1962. A method for the estimation of serum creatine kinase and its use in comparing creatine kinase and aldolase activity in normal and pathological sera. Clin Chim Acta 7: 597-603.
- HWANG C, SINSKEY AJ AND LODISH HF. 1992. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257: 1496-1502.
- JANSSEN E, TERZIC A, WIERINGA B AND DZEJA PP. 2003. Impaired intracellular energetic communication in muscles from creatine kinase and adenylate kinase (M-CK/AK1) double knock-out mice. J Biol Chem 278: 30441-30449.
- JONES DP. 2006. Redefining oxidative stress. Antioxid Redox Signal 8: 1865-1879.
- JONES DP, GO YM, ANDERSON CL, ZIEGLER TR, KINKADE JR JM AND KIRLIN WG. 2004. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J 18: 1246-1248.
- LAUBE GF, SHAH V, STEWART VC, HARGREAVES IP, HAQ MR, HEALES SJR AND VAN’T HOFF WG. 2006. Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr Nephrol 21: 503-509.
- LEECH NL, BARRETT KC AND MORGAN GA. 2005. SPSS for intermediate statistics. Use and interpretation, 2nd ed., London, England: Lawrence Erlbaum Associates Publishers.
- LEONG SF, LAI JC, LIM L AND CLARK JB. 1981. Energy-metabolising enzymes in brain regions of adult and aging rats. J Neurochem 37: 1548-1556.
- LEVTCHENKO EN, WILMER MJ, JANSSEN AJ, KOENDERINK JB, VISCH HJ, WILLEMS PH, DE GRAAF-HESS A, BLOM HJ, VAN DEN HEUVEL HP AND MONNENS LA. 2006. Decreased intracellular, ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res 59: 287-292.
- LOWRY OH, ROSEBROUGH NJ AND FARR AL. 1951. Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275.
- MORIARTY-CRAIGE SE AND JONES DP. 2004. Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr 24: 481-509.
- NASRALLAH F, FEKI M AND KAABACHI N. 2010. Creatine and creatine deficiency syndromes: biochemical and clinical aspects. Pediatr Nephrol 42: 163-171.
- PACKER L AND CADENAS E. 2011. Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J Clin Biochem Nutr 48: 26-32.
- PARK MA, PEJOVIC V, KERISIT KG, JUNIUS S AND THOENE JG. 2006. Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase C delta. J Am Soc Nephrol 17: 3167-3175.
- PARK MA AND THOENE JG. 2005. Potential role of apoptosis in development of the cystinotic phenotype. Pediatr Nephrol 20: 441-446.
- PEREIRA OLIVEIRA PR, RODRIGUES-JUNIOR V, RECH VC AND WANNMACHER CM. 2007. Cystine inhibits creatine kinase activity in pig retina. Arch Med Res 38: 164-169.
- PRICES NC, COHN M AND SCHIRMER RH. 1975. Fluorescent and Spin Label Probes of the Environments of the Sulfhydryl Groups of Porcine Muscle Adenylate Kinase. J Biol Chem 250: 644-652.
- RECH VC, ATHAYDES GA, FEKSA LR, DORNELLES PK, RODRIGUES-JUNIOR V, DUTRA-FILHO CS, DE SOUZA WYSE AT, WAJNER M AND WANNMACHER CM. 2006. Inhibition of creatine kinase activity by cystine in the kidney of young rats. Pediatr Res 1124: 190-195.
- RECH VC, FEKSA LR, FLECK RM, ATHAYDES GA, DORNELLES PK, RODRIGUES-JUNIOR V AND WANNMACHER CM. 2008. Cysteamine prevents inhibition of thiol-containing enzymes caused by cystine or cystine dimethyl ester loading in rat brain cortex. Metab Brain Dis 23: 133-145.
- SALMON RF AND BAUM M. 1990. Intracellular cystine loading inhibits transport in the rabbit proximal convoluted tubule. J Clin Invest 85: 340-344.
- SENGUPTA P. 2013. The Laboratory Rat: Relating Its Age With Human’s. Int J Prev Med 4: 624-630.
- WALLIMANN T, WYSS M, BRDICZKA D, NICOLAY K AND EPPENBERGER HM. 1992. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J 281: 21-40.
- WALTERS DW AND GILBERT HF. 1986. Thiol/disulfide redox equilibrium and kinetic behavior of chicken liver fatty acid synthase. J Biol Chem 261: 13135-13143.
- WILKINSON B AND GILBERT HF. 2004. Protein disulfide isomerase. Biochim Biophys Acta 1699: 35-44.
- WILMER MJ, KLUIJTMANS LA, VAN DER VELDEN TJ, WILLEMS PH, SCHEFFER PG, MASEREEUW R, MONNENS LA, VAN DEN HEUVEL LP AND LEVTCHENKO EN. 2011. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim Biophys Acta 1812: 643-651.
- WILMER MJ, WILLEMS PH, VERKAART S, VISCH HJ, DE GRAAF-HESS A, BLOM HJ, MONNENS LA, VAN DEN HEUVEL LP AND LEVTCHENKO EN. 2007. Cystine dimethylester model of cystinosis: still reliable? Pediatr Res 62: 151-155.
- ZAHLER WL AND CLELAND WW. 1968. A specific and sensitive assay for disulfides. J Biol Chem 243: 716-719.
Publication Dates
-
Publication in this collection
07 Dec 2017 -
Date of issue
Jan-Mar 2018
History
-
Received
10 June 2016 -
Accepted
08 Aug 2016