Resumos
Relatamos um caso de paciente com Síndrome do Olho de Gato (Cat Eye Syndrome-CES) e interrupção do arco aórtico tipo B, um achado típico na síndrome da deleção 22q11.2. A análise cromossômica e a técnica de hibridização fluorescente in situ (FISH) mostraram um cromossomo marcador isodicêntrico supranumerário com bi-satélite derivado do cromossomo 22. O segmento de 22pter a 22q11.2 no cromossomo supranumerário encontrado em nosso paciente não estava em sobreposição com a região deletada em pacientes com a síndrome da deleção 22q11.2. Entretanto, o achado de interrupção do arco aórtico tipo B não é usual na CES, mas é um defeito cardíaco freqüente na síndrome da deleção 22q11.
Aorta torácica; síndrome do Olho de Gato; deleção cromossômica; cardiopatias congênitas
Informamos un caso de paciente con Síndrome de Ojo de Gato (Cat Eye Syndrome-CES) e Interrupción del Arco Aórtico tipo B, un hallazgo típico en el síndrome de la deleción 22q11.2. El análisis cromosómico y la técnica de hibridación in situ fluorescente (FISH) mostraron un cromosoma marcador isodicéntrico supernumerario bisatelitado derivado del cromosoma 22. El segmento de 22pter a 22q11.2 en el cromosoma supernumerario encontrado en nuestro paciente no estaba en sobreposición con la región deletada en pacientes con el síndrome de la deleción 22q11.2. Con todo, el hallazgo de interrupción del arco aórtico tipo B no es usual en el CES, sino que es un defecto cardíaco frecuente en el síndrome de deleción 22q11.
Aorta torácica; Síndrome de Ojo de Gato; deleción cromosómica; cardiopatías congénitas
We report a patient with cat eye syndrome and interrupted aortic arch type B, a typical finding in the 22q11.2 deletion syndrome. Chromosomal analysis and fluorescent in situ hybridization (FISH) showed a supernumerary bisatellited isodicentric marker chromosome derived from chromosome 22. The segment from 22pter to 22q11.2 in the supernumerary chromosome found in our patient does not overlap with the region deleted in patients with the 22q11.2 deletion syndrome. However, the finding of an interrupted aortic arch type B is unusual in CES, although it is a frequent heart defect in the 22q11 deletion syndrome.
Aorta, thoracic; Cat Eye Syndrome; chromosome deletion; heart defects, congenital
RELATO DE CASO
Interrupção do arco aórtico tipo B em uma paciente com Síndrome de Olho de Gato
Sintia Iole Nogueira BelangeroI; Fernanda Teixeira da Silva BelluccoI; Mirlene C. S. P. CernachI; April M. HackerII; Beverly S. EmanuelII; Maria Isabel MelaragnoI
IUniversidade Federal de São Paulo, São Paulo, SP - Brasil
IIChildren´s Hospital of Philadelphia (University of Pennsylvania), Filadélfia, Pensilvânia - Estados Unidos
Correspondência Correspondência: Sintia Iole Nogueira Belangero Rua Botucatu, 740 - Vila Clementino 04023-900 - São Paulo, SP, Brasil E-mail: sintia.morf@epm.br
RESUMO
Relatamos um caso de paciente com Síndrome do Olho de Gato (Cat Eye Syndrome-CES) e interrupção do arco aórtico tipo B, um achado típico na síndrome da deleção 22q11.2. A análise cromossômica e a técnica de hibridização fluorescente in situ (FISH) mostraram um cromossomo marcador isodicêntrico supranumerário com bi-satélite derivado do cromossomo 22. O segmento de 22pter a 22q11.2 no cromossomo supranumerário encontrado em nosso paciente não estava em sobreposição com a região deletada em pacientes com a síndrome da deleção 22q11.2. Entretanto, o achado de interrupção do arco aórtico tipo B não é usual na CES, mas é um defeito cardíaco freqüente na síndrome da deleção 22q11.
Palavras-chave: Aorta torácica, síndrome do Olho de Gato, deleção cromossômica, cardiopatias congênitas.
Introdução
A Síndrome do Olho de Gato (Cat Eye Syndrome - CES; OMIM #115470) é uma síndrome rara clinicamente caracterizada por fissuras palpebrais oblíquas direcionadas para baixo, coloboma ocular, malformações anorretais, cardíacas e renais, depressões ou pregas pré-auriculares e retardo mental1,2. A CES é freqüentemente associada com variabilidade fenotípica significante, variando de pacientes com fenótipo quase normal àqueles com anormalidades graves1. Nenhuma das características está presente de forma consistente. Apenas 41% dos pacientes com CES apresentam uma combinação clássica de coloboma de íris, anomalias anais e anomalias pré-auriculares1. Dessa forma, muitos pacientes não podem ser identificados como portadores de CES somente através do fenótipo. O diagnóstico é baseado na presença de um cromossomo marcador supranumerário, derivado do cromossomo 22. Esse cromossomo marcador é usualmente isodicêntrico, contem um único centrômero ativo, e freqüentemente exibe satélites em ambas as extremidades. Assim, os pacientes apresentam tetrassomia parcial de 22-pter a 22q11.23. Os pontos de quebra moleculares que originam o cromossomo da CES (CEC) usualmente estão em clusters em dois intervalos. O ponto de quebra proximal é o mais comum e corresponde ao intervalo do ponto de quebra de deleção proximal encontrado para a deleção 22q11.
O ponto de quebra mais distal se sobrepõe ao ponto de quebra de deleção distal comum da deleção de 3 Mb visto na síndrome de deleção 22q114. Assim, com base na localização dos dois pontos de quebra, o CEC pode ser classificado em três tipos: eles podem ser isodicêntricos com ambos os pontos de quebra proximais ou ambos distais, resultando em um cromossomo menor, ou um cromossomo simétrico maior, respectivamente. O CEC também pode ser originado através de um ponto de quebra proximal e um distal, resultando em um CEC assimétrico. Nesse último caso, há uma cópia adicional da região cromossômica DiGeorge (DGCR), enquanto no CEC maior isodicêntrico há duas, e no CEC menor não há uma cópia extra da DGCR.
A fronteira distal da região cromossômica da CES é proximal à DGCR5. Ela cobre aproximadamente 2 Mb de 22q11.2 com sua fronteira distal. Não foi encontrada nenhuma correlação óbvia entre a gravidade do fenótipo associado e o tamanho da região duplicada na CES4,5. No presente estudo, descrevemos um paciente com um cromossomo marcador supranumerário, síndrome do olho de gato e interrupção do arco aórtico tipo B.
Relato de caso
Paciente
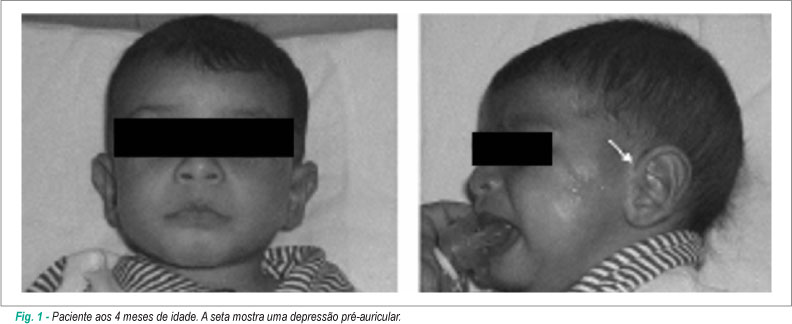
O Comitê de Ética em Pesquisa da Universidade Federal de São Paulo (UNIFESP) aprovou esse estudo e os pais do paciente deram seu consentimento informado. O paciente era do sexo feminino e a primeira filha de pais jovens (mãe 22 e pai 26 anos). O parto foi cesáreo com 39 semanas de gestação. O peso ao nascer foi 2760 g, comprimento 47 cm, e circunferência cefálica, 34 cm. A mãe observou que a criança parecia cansada após alimentar-se. Aos três meses de idade, um ecocardiograma foi realizado e mostrou comunicação interventricular (CIV), hipoplasia moderada do anel e válvula aórticos, dilatação moderada do tronco pulmonar, arco aórtico voltada para a esquerda, presença de interrupção do arco aórtico tipo B e presença de um canal arterial de bom calibre com shunt arterial aórtico-pulmonar.Esses achados foram posteriormente confirmados por outros ecocardiogramas. Aos 4 meses de idade (Figura 1), o peso era 4410 g (<3%ile), comprimento 61.5 cm (50%ile) e a circunferência cefálica era 41 cm (50%ile). A criança apresentava uma testa larga e proeminente, fissuras palpebrais oblíquas direcionadas para baixo, hipertelorismo, pregas epicânticas internas, depressões preauriculares bilaterais, orelhas anormais, ponte nasal achatada, anteversão das narinas, filtrum longo, boca de carpa e hipoplasia malar discreta. A criança faleceu aos 5 meses de idade. A causa da morte foi uma infecção (broncopneumonia) que não permitiu a correção cirúrgica da cardiopatia.
Análise citogenética
A análise cromossômica revelou um cromossomo marcador supranumerário com bi-satélite em todas as metáfases analisadas. Os cariótipos dos pais foram normais. A técnica de FISH usando a sonda centromérica 14,22 (D14Z1/D22Z1) (Vysis®) foi positiva no cromossomo marcador. A hibridização com a sonda DiGeorge/VCFS-Tuple1 (Cytocell®) mostrou ausência da região cromossômica DGS/VCFS no cromossomo marcador. A técnica FISH com sondas cosmid mostrou que o cromossomo marcador hibridizava com a sonda c106E4 (proximal), mas não com a c103A2 (distal). Esse fato identificou o marcador como um cromossomo 22 isodicêntrico com ponto de quebra na região de baixa repetição de cópia A (LCR A) (Figura 2).
Discussão
Considerando o fenótipo da criança e que aproximadamente 50% dos casos com interrupção do arco aórtico tipo B estão associados com a microdeleção de 22q11.2, o diagnóstico inicial da paciente foi a síndrome da deleção de 22q11.26-8. Entretanto, a paciente apresentava um cariótipo com um cromossomo supranumerário e foi diagnosticada como sendo portadora de CES. Uma revisão de 74 pacientes com CES mostrou a presença de defeitos cardíacos em 37, dos quais 16 tinham um retorno venoso pulmonar anômalo total e três tinham Tetralogia de Fallot. Outras anomalias cardíacas incluíam defeito do septo atrial, hipoplasia da válvula mitral, átrio ou ventrículo, monoventrículo e persistência da veia cava superior esquerda1.
Outro estudo relatou que 51 pacientes com CES tinham defeitos cardíacos congênitos2. Dezoito deles tinham defeito do septo ventricular, 15 tinham conexão venosa-pulmonar anômala, 16 tinham defeito do septo, 7 tinham Tetralogia de Fallot, 8 pacientes tinham persistência do canal arterial, 5 tinham malformações aórticas, 5 tinham estenose pulmonar, 3 tinham atresia de tricúspide e 3 tinham síndrome do coração esquerdo hipoplásico.
Nossa paciente apresentou um cromossomo CES simétrico que não continha a região deletada em pacientes com a síndrome da deleção 22q11.2. Estudos anteriores indicaram que a maioria (6/9) das duplicações cromossômicas da CES não se estendiam à região comumente deletada de pacientes com a síndrome da deleção 22q11.25. Entretanto, nossa paciente apresentava interrupção do arco aórtico tipo B, um defeito cardíaco freqüente na síndrome da deleção 22q11.2, mas ainda não descrita na CES. Assim, embora a etiologia para essa lesão cardíaca seja provavelmente o resultado de uma anormalidade cromossômica, outras etiologias potenciais, como as outras regiões genômicas envolvidas, etiologias ou interações ambientais em ter fatores genéticos e ambientais deverão ser exploradas.
Agradecimentos
Esse trabalho recebeu apoio financeiro do CNPq e CAPES, Brasil e fundos DC02027, CA39926 do NIH (BSE) e de Charles E.H. Upham endowed chair (BSE).
Potencial Conflito de Interesses
Declaro não haver conflito de interesses pertinentes.
Fontes de Financiamento
O presente estudo foi financiado por CNPq, CAPES e DC02027, CA39926 (Grants) de NIH (BSE) e de Charles E. H. Uphan endowed chair.
Vinculação Acadêmica
Este artigo é parte de tese de Doutorado de Sintia Iole Nogueira Belangero pela Universidade Federal de São Paulo.
Artigo recebido em 21/09/07, revisado recebido em 05/12/07; aceito em 12/02/08.
- 1. Berends MJ, Tan-Sindhunata G, Leegte B, van Essen AJ. Phenotypic variability of Cat-Eye syndrome. Genet Couns. 2001; 12: 23-34.
- 2. Rosias PR, Sijstermans JM, Theunissen PM, Pulles-Heintzberger CF, De Die-Smulders CE, Engelen JJ, et al. Phenotypic variability of the cat eye syndrome: case report and review of the literature. Genet Couns. 2001; 12: 273-82.
- 3. Mears AJ, el-Shanti H, Murray JC, McDermid HE, Patil SR. Minute supernumerary ring chromosome 22 associated with cat eye syndrome: further delineation of the critical region. Am J Hum Genet. 1995; 57: 667-73.
- 4. McTaggart KE, Budarf ML, Driscoll DA, Emanuel BS, Ferreira P, McDermid HE. Cat eye syndrome chromosome breakpoint clustering: identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenet Cell Genet. 1998; 81: 222-8.
- 5. Mears AJ, Duncan AM, Budarf ML, Emanuel BS, Sellinger B, Siegel-Burtelt J, et al. Molecular characterization of the marker chromosome associated with cat eye syndrome. Am J Hum Genet. 1994; 55: 134-42.
- 6. Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003; 112: 101-7.
- 7. Takahashi K, Kuwahara T, Nagatsu M. Interruption of the aortic arch at the isthmus with DiGeorge syndrome and 22q11.2 deletion. Cardiol Young. 1999; 9: 516-8.
- 8. Goldmuntz E, Clark B, Mitchell L, Jawad AF, Cuneo BF, Reed L. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998; 32: 492-8.
- 9. Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000; 9: 489-501.
Correspondência:
Datas de Publicação
-
Publicação nesta coleção
20 Jul 2009 -
Data do Fascículo
Maio 2009
Histórico
-
Recebido
21 Set 2007 -
Revisado
05 Dez 2007 -
Aceito
12 Fev 2008