Abstracts
Dowling-Degos disease (DDD) is a rare genetic disease of the skin (reticulate pigmented anomaly), clinically characterized by flexural brown pigmented reticulate macules, comedo-like papules on the back, neck and pitted perioral or facial scars. We present the case of a 51 year-old man with macrocomedo-like lesions, pitted scars, cysts, hyperpigmented macules in his back, chest, axillae, neck, groin and face. The patient reported having two children, three brothers and a father with a similar condition. The histopathology of the skin biopsies was very characteristic of Dowling-Degos disease, showing dilated follicular, fingerlike projections called rete ridges (dermal pegs), with thinning of the suprapapillary plates, resulting in an "antler-like" pattern and increased pigmentation of the basal layer.
Hyperpigmentation; Keratin-5; Skin diseases; genetic
A doença de Dowling-Degos é uma genodermatose rara que consiste numa desordem pigmentar reticulada. Caracteriza-se pela presença de máculas hiperpigmentadas nas regiões flexurais com distribuição em rede; lesões tipo comedão no dorso e na região cervical; e cicatrizes cribriformes na face, particularmente periorais. Apresentamos um caso de um paciente de 51 anos, masculino, com lesões tipo macrocomedões, cicatrizes cribriformes, cistos e máculas hipercrômicas no dorso, tórax anterior, axilas, pescoço, região genital e face. Relatava ter dois filhos, três irmãos e o pai com quadro semelhante. As biópsias de pele foram características da doença de Dowling-Degos, mostrando dilatação folicular, epiderme digitiforme, com áreas de aspecto de "chifre de veado" e focos de hiperpigmentação da camada basal.
Dermatopatias genéticas; Hiperpigmentação; Queratina-5
DERMATOPATOLOGIA
Dowling-Degos disease: classic clinical and histopathological presentation*
Carolina Cotta ZimmermannI; Deborah SforzaII; Priscila Marques de MacedoII; Luna Azulay-AbulafiaIII; Maria de Fatima G. S. AlvesIII; Sueli Coelho da S. CarneiroIII
IMD Resident in Dermatology, Pedro Ernesto University Hospital, State University of Rio de Janeiro (UERJ), Rio de Janeiro (RJ), Brazil
IIMD, PhD in Dermatology, Pedro Ernesto University Hospital, State University of Rio de Janeiro (UERJ), Rio de Janeiro (RJ), Brazil IIIMD, PhD and Associate Professor at the State University of Rio de Janeiro (UERJ), Rio de Janeiro (RJ), Brazil
Mailing address
ABSTRACT
Dowling-Degos disease (DDD) is a rare genetic disease of the skin (reticulate pigmented anomaly), clinically characterized by flexural brown pigmented reticulate macules, comedo-like papules on the back, neck and pitted perioral or facial scars. We present the case of a 51 year-old man with macrocomedo-like lesions, pitted scars, cysts, hyperpigmented macules in his back, chest, axillae, neck, groin and face. The patient reported having two children, three brothers and a father with a similar condition. The histopathology of the skin biopsies was very characteristic of Dowling-Degos disease, showing dilated follicular, fingerlike projections called rete ridges (dermal pegs), with thinning of the suprapapillary plates, resulting in an "antler-like" pattern and increased pigmentation of the basal layer.
Keywords: Hyperpigmentation; Keratin-5; Skin diseases, genetic
INTRODUCTION
Reticulated hyperpigmentation anomalies were initially distinguished from acanthosis nigricans by Dowling and Freudenthal in 1938 and designated Dowling-Degos Disease (DDD) by Wilson-Jones and Grice in 1978. 1
The onset of the disease - a rare genodermatosis of autosomal dominant transmission with variable penetrance - usually occurs in the third or fourth decade of life. It is also referred to as 'Dark Dot Disease'. 2 It affects predominantly women in a ratio of 2:1 and has no race predilection. 3 A recent study involving six people with the disease belonging to two different families revealed mutation in the keratin 5 gene (KRT5), highlighting the importance of keratin 5 in cell adhesion and in the transport and transfer of melanosomes. 4
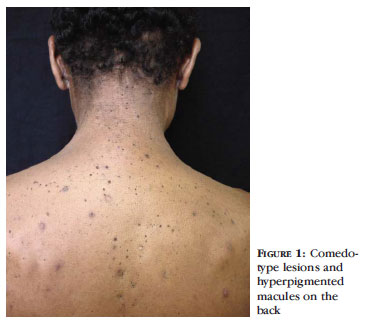
The disease is characterized by acquired reticulated skin hyperpigmentation, which begins in the armpits, groin and later spreads to other skin folds. It can affect (although more uncommonly), the wrist, the antecubital and popliteal hollow, the face, scalp, scrotum and vulva . 1 Some patients complain of itching in the macula. These lesions do not change with sun exposure. Comedo-type black lesions on the face, back and in the same areas described above can also be observed 3, as well as cribriform scars and perioral acne in patients with no history of acne. Other features may be present such as mental retardation and pillar cyst. 2 While at the benign stage the disease shows no inflammatory phenomena, it is nevertheless very unsightly. 3
Histopathology and the clinical picture are both very characteristic of the disease and allow a conclusive diagnosis.
CASE REPORT
Male patient, aged 51, born in Rio de Janeiro, complained of papules on the back, neck, armpits and genitalia which drained serous purulent and serohemorrhagic exudate, with spontaneous resolution and recurrence since childhood, with worsening over the last 3 years (Figures 1 and 2). The patient reported a similar clinical condition of his late father, his three brothers and two of his sons. For a variety of reasons these relatives had not been submitted to clinical evaluation.
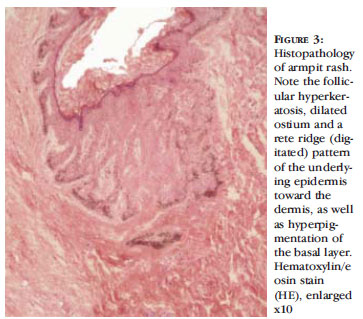
Biopsies were performed for three different types of lesions: hyperchromic macule in the armpit, the macrocomedones and the cribriform scar on the back. All showed essentially the same histopathological finding. Follicular hyperkeratosis was observed, with dilation of the ostium, a rete ridge (digitated) pattern of the underlying epidermis toward the dermis, and hyperpigmentation of the basal layer (Figures 3, 4 and 5). In the biopsy of the cribriform scar, pseudocysts were also observed (Figure 5). The outcome of the clinical and histopathological tests pointed conclusively to Dowling-Degos disease.
DISCUSSION
DDD is a late-onset genodermatosis, occurring usually in adulthood, with the appearance of hyperpigmented macules. The pigmentation gradually increases over time. The disease initially affects the armpits and groin and later the intergluteal, inframammary regions and the neck and trunk. Pruritus in the folds may be reported. Over time comedo-like lesions also appear on the back and neck, and perioral cribriform scars and follicular cysts. The disease has been associated with Hidradenitis Suppurativa (HS), also known as 'Acne Inversa', and multiple keratoacanthomas, perhaps because DDD causes a defect in the phylo sebaceous epithelial proliferation. 5.6 In the case of our patient the lesions had been present since prepuberty.
Histopathological findings of DDD include moderate orthokeratosis or hyperkeratosis, thinning of the suprapapillary epithelium and elongation of the papillae with basal layer hyperpigmentation. These threadlike growths of the epidermis have the appear-ance of "antlers" and generally involve the follicle with a follicular plug. A perivascular lymphohistiocytic infiltrate in the papillary dermis and pseudo-horny cysts can also be observed. All the biopsies from our patient conformed to these findings. Staining for S100 does not generally show increased numbers of melanocytes, suggesting that the pigment does not arise from increased density of melanocytes. 1 It was not however possible to perform this test.
Some authors refer to Acropigmentation of Kitamura, Haber's disease and Galli-Galli disease as differential diagnoses of DDD. 2.5
Reticulate Acropigmentation of Kitamura (RAPK) is a sporadic autosomal dominant disease of unknown origin. It prevails in Japan, but has been observed worldwide. Its clinical features consist of hyperpigmented atrophic macules on the backs of the hands and feet. Its onset is in childhood. The lesions darken with time and worsen with sun exposure. Pitting on the palms and soles and dorsa of the fingers can also be found. 2.5
Harber's disease is characterized by a photosensitive facial rosacea-like rash which develops in adolescence, progressing later to seborrheic keratosis-like papules, comedones, cribriform scars, reticulate hyperpigmentation of the trunk, proximal extremities and armpits. 7.8
Galli-Galli disease is an acantholytic variant of Dowling-Degos disease which presents in people aged between 15 and 56. 9 While the mechanism of acantholysis is unknown authors have suggested that it results from of loss of epidermal cell stability due to a keratin 5 defect. Clinical symptoms include the presence of hyperpigmentation of the flexures together with itching, and sometimes with erythematous, scaly papules on these sites as well as on the trunk and proximal extremities. The histopathology resembles that of DDD, but with foci of acantholysis. Another differential diagnosis with Galli-Galli disease would be epidermolysis bullosa with mottled pigmentation, although this disease has other additional features such as blisters and palmoplantar hyperkeratosis. Treatment usually consists of topical steroids and retinoids, with little success. 10
DDD must also be differentiated from acanthosis nigricans which also shows a predilection for skin folds, but with the emergence of velvety plaques. Histopathologically the papillae are less elongated and without follicular involvement. Patients with neurofibromatosis may also have freckle-like (ephelides) lesions in the armpits, but this disease is easily distinguished from DDD. 3
No other complementary examination exists to aid diagnosis of DDD apart from a typical histopathologic examination associated with compatible clinical features, as we did in this case.
A variety of treatments can be tried but with unsatisfactory outcomes: topical hydroquinone, tretinoin, adapalene and corticosteroids, as well as Er:YAG lasers11. Isotretinoin would be an option since it produces an alteration in the pattern of keratinization. In the literature we found a report of three patients receiving 20mg/day of oral and topical isotretinoin. However the treatment was ineffective and oral therapy was discontinued, retaining only the topical retinoid. 12.13

REFERENCES
- 1. Kim YC, Davis MD, Schanbacher CF, Su WP. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999;40:462-7.
- 2. Schnur RE, Heymann WR. Reticulate Hyperpigmentation. Semin Cutan Med Surg. 1997;16:72-80.
- 3. Azulay-Abulafia L, Porto JA, Souza MAJ, Wrobel R, Brito MA, Valverde RV. Doença de Dowling-Degos. An Bras Dermatol. 1992;67:275-8.
- 4. Betz RC, Planko L, Eigelshoven S, Hanneken S, Pasternack SM, Bussow H, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-9.
- 5. Chang MW. Disorders of Hyperpigmentation. In: Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. London: Mosby Elsevier; 2008. p.1820-5.
- 6. Fenske NA, Groover CE, Lober CW, Espinoza CG. Dowling-Degos disease, hidradenitis suppurativa, and multiple keratoacanthomas: a disorder that may be caused by a single underlying defect in pilsebaceous epithelial proliferation. J Am Acad Dermatol. 1991;24:888-92.
- 7. McCormack CJ, Cowen P. Harber's syndrome. Australas J Dermatol. 1997;38:82-4.
- 8. Nishizawa A, Nakano H, Satoh T, Takayama K, Sawamura D, Yokozeki H. Harber's syndrome may be a clinical entity different from Dowling-Degos disease. Br J Dermatol. 2009;160:215-7.
- 9. Braun-Falco M, Volgger W, Borelli S, Ring J, Disch R. Galli-Galli disease: an unrecognized entity or an acantholytic variant of Dowling-Degos disease? J Am Acad Dermatol. 2001;45:760-3.
- 10. Gilchrist H, Jackson S, Morse L, Nicotri T, Nesbitt LT. Galli-Galli disease: A case report with review of the literature. J Am Acad Dermatol. 2008;58:299-302.
- 11. Wenzel G, Petrow W, Tappe K, Gerdsen R, Uerlich WP, Bieber T. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2,5 years. Dermatol Surg. 2003;29:1161-2.
- 12. Bhagwat PV, Tophakhane RS, Shashikumar BM, Noronha TM, Naidu V. Three cases of Dowling Degos disease in two families. Indian J Dermatol Venereol Leprol. 2009;75:398-400.
- 13. Oppolzer G, Schwarz T, Duschet P, Brenner W, Gschnait F. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids. Hautarzt. 1987;38:615-8.
Publication Dates
-
Publication in this collection
01 Dec 2011 -
Date of issue
Oct 2011
History
-
Received
01 Aug 2010 -
Accepted
31 Aug 2010