Abstracts
The hypophosphatemic conditions that interfere in bone mineralization comprise many hereditary or acquired diseases, all of them sharing the same pathophysiologic mechanism: reduction in the phosphate reabsorption by the renal tubuli. This process leads to chronic hyperphosphaturia and hypophosphatemia, associated with inappropriately normal or low levels of calcitriol, causing osteomalacia or rickets in children and osteomalacia in adults. X-linked hypophosphatemic rickets, autosomal-dominant hypophosphatemic rickets, and tumor-induced osteomalacia are the main syndromes involved in the hypophosphatemic rickets. Although these conditions exhibit different etiologies, there is a common link among them: increased activity of a phosphaturic factor, being the fibroblast growth factor 23 (FGF-23) the most studied one and to which is attributed a central role in the pathophysiology of the hyperphosphaturic disturbances. Activating mutations of FGF-23 and inactivating mutations in the PHEX gene (a gene on the X chromosome that codes for a Zn-metaloendopeptidase proteolytic enzyme which regulates the phosphate) involved in the regulation of FGF-23 have been identified and have been implicated in the pathogenesis of these disturbances. Genetic studies tend to show that the phosphorus homeostasis depends on a complex osteo-renal metabolic axis, whose mechanisms of interaction have been poorly understood so far. This paper reviews the current knowledge status concerning the pathophysiology of phosphate metabolism regulation and the pathophysiologic basis of hypophosphatemic rickets. It also analyzes the clinical picture and the therapeutic aspects of these conditions as well.
Hypophosphatemic rickets; Osteomalacia; FGF-23; PHEX gene; Phosphate metabolism
Os distúrbios hipofosfatêmicos que comprometem a mineralização óssea englobam várias doenças, hereditárias e adquiridas, as quais compartilham um mesmo mecanismo fisiopatológico: a diminuição da reabsorção de fosfato nos túbulos renais. Este processo promove hiperfosfatúria e hipofosfatemia crônicas, associadas a níveis inapropriadamente normais ou baixos de 1,25 (OH)2D3, com conseqüente desordem do metabolismo ósteo-mineral, resultando em raquitismo e osteomalácia na faixa etária pediátrica e em osteomalácia nos adultos. O raquitismo hipofosfatêmico ligado ao X, o raquitismo hipofosfatêmico autossômico dominante e a osteomalácia induzida por tumor são as principais síndromes que constituem os raquitismos hipofosfatêmicos. Apesar de estas doenças apresentarem etiopatogenias distintas, as evidências bioquímico-moleculares indicam uma base fisiopatológica em comum: maior atividade de um agente fosfatúrico, sendo o fator de crescimento do fibroblasto 23 (FGF-23) o mais estudado e ao qual é atribuído um papel central na fisiopatologia destes distúrbios. Várias mutações ativadoras do gene do FGF-23 e mutações inativadoras do gene localizado no cromossomo X que codifica uma enzima proteolítica Zn-metaloendopeptidase reguladora do fosfato (PHEX), implicada na regulação do FGF-23, já foram identificadas, e sua participação reconhecida na gênese destes distúrbios. Os dados dos estudos genéticos nesta área convergem para a hipótese de que a homeostase do fósforo estaria vinculada a um complexo eixo metabólico ósteo-renal, cujos mecanismos de interação entre seus vários componentes têm sido aos poucos elucidados. Este artigo revisa o atual estado de conhecimento dos mecanismos fisiológicos envolvidos na regulação do metabolismo do fosfato, das bases fisiopatológicas dos raquitismos hipofosfatêmicos e analisa aspectos clínicos e de tratamento disponíveis para estas condições.
Raquitismo hipofosfatêmico; Osteomalácia; FGF-23; Gene PHEX; Metabolismo do fosfato
ORIGINAL ARTICLE
Hypophosphatemic rickets and osteomalacia
Raquitismo hipofosfatêmico e osteomalácia
Hamilton de Menezes FilhoI; Luiz Claudio G. de CastroII; Durval DamianiI
IPediatric Endocrinology Unit, Instituto da Criança Hospital das Clínicas, São Paulo University Medical School USP, São Paulo, SP
IIPediatric Endocrinology Unit, Department of Pediatrics, Medicine College, Brasília University UnB, Brasília, DF, Brazil
Address for correspondence Address for correspondence: Durval Damiani Rua Bela Cintra 2117, apto. 9 01415-002 São Paulo, SP E-mail: durvald@iconet.com.br
ABSTRACT
The hypophosphatemic conditions that interfere in bone mineralization comprise many hereditary or acquired diseases, all of them sharing the same pathophysiologic mechanism: reduction in the phosphate reabsorption by the renal tubuli. This process leads to chronic hyperphosphaturia and hypophosphatemia, associated with inappropriately normal or low levels of calcitriol, causing osteomalacia or rickets in children and osteomalacia in adults. X-linked hypophosphatemic rickets, autosomal-dominant hypophosphatemic rickets, and tumor-induced osteomalacia are the main syndromes involved in the hypophosphatemic rickets. Although these conditions exhibit different etiologies, there is a common link among them: increased activity of a phosphaturic factor, being the fibroblast growth factor 23 (FGF-23) the most studied one and to which is attributed a central role in the pathophysiology of the hyperphosphaturic disturbances. Activating mutations of FGF-23 and inactivating mutations in the PHEX gene (a gene on the X chromosome that codes for a Zn-metaloendopeptidase proteolytic enzyme which regulates the phosphate) involved in the regulation of FGF-23 have been identified and have been implicated in the pathogenesis of these disturbances. Genetic studies tend to show that the phosphorus homeostasis depends on a complex osteo-renal metabolic axis, whose mechanisms of interaction have been poorly understood so far. This paper reviews the current knowledge status concerning the pathophysiology of phosphate metabolism regulation and the pathophysiologic basis of hypophosphatemic rickets. It also analyzes the clinical picture and the therapeutic aspects of these conditions as well.
Keywords: Hypophosphatemic rickets; Osteomalacia; FGF-23; PHEX gene; Phosphate metabolism
RESUMO
Os distúrbios hipofosfatêmicos que comprometem a mineralização óssea englobam várias doenças, hereditárias e adquiridas, as quais compartilham um mesmo mecanismo fisiopatológico: a diminuição da reabsorção de fosfato nos túbulos renais. Este processo promove hiperfosfatúria e hipofosfatemia crônicas, associadas a níveis inapropriadamente normais ou baixos de 1,25 (OH)2D3, com conseqüente desordem do metabolismo ósteo-mineral, resultando em raquitismo e osteomalácia na faixa etária pediátrica e em osteomalácia nos adultos. O raquitismo hipofosfatêmico ligado ao X, o raquitismo hipofosfatêmico autossômico dominante e a osteomalácia induzida por tumor são as principais síndromes que constituem os raquitismos hipofosfatêmicos. Apesar de estas doenças apresentarem etiopatogenias distintas, as evidências bioquímico-moleculares indicam uma base fisiopatológica em comum: maior atividade de um agente fosfatúrico, sendo o fator de crescimento do fibroblasto 23 (FGF-23) o mais estudado e ao qual é atribuído um papel central na fisiopatologia destes distúrbios. Várias mutações ativadoras do gene do FGF-23 e mutações inativadoras do gene localizado no cromossomo X que codifica uma enzima proteolítica Zn-metaloendopeptidase reguladora do fosfato (PHEX), implicada na regulação do FGF-23, já foram identificadas, e sua participação reconhecida na gênese destes distúrbios. Os dados dos estudos genéticos nesta área convergem para a hipótese de que a homeostase do fósforo estaria vinculada a um complexo eixo metabólico ósteo-renal, cujos mecanismos de interação entre seus vários componentes têm sido aos poucos elucidados. Este artigo revisa o atual estado de conhecimento dos mecanismos fisiológicos envolvidos na regulação do metabolismo do fosfato, das bases fisiopatológicas dos raquitismos hipofosfatêmicos e analisa aspectos clínicos e de tratamento disponíveis para estas condições.
Descritores: Raquitismo hipofosfatêmico; Osteomalácia; FGF-23; Gene PHEX; Metabolismo do fosfato
THE BONE TISSUE IS A HIGHLY dynamic structure throughout life. It has well known metabolic and mechanical functions, which are essential for the body homeostasis. Bone-mineral metabolism is coordinated by an intricate physiological network and reflects a continuous interaction between genetics and environment, with ontogenetic particularities in each development stage. During fetal life, childhood and adolescence bone-mineral metabolism moves toward bone formation and modeling, promoting skeleton growth through changes in bone dimensions, geometry and density. Meanwhile, bone-remodeling process occurs continuously throughout life and consists in substitution of old bone by new one. This renewal process is elementary to maintain the bone resistance and quality and for the integrity of the body physiology as well, since bone is an ion reservoir tissue, mainly for calcium and phosphorus, critical elements for the systemic electrolytic equilibrium.
Bone-mineral homeostasis is complex and some of its pathways have not been completely elucidated yet. The parathyroid hormone (PTH)-calcitonin-1,25(OH)2Vitamin D3 (calcitriol) axis plays a fundamental role in this bone-mineral metabolism, but it could not respond alone for the whole dynamics of this equilibrium (1). The ascertainment of other involved regulatory biochemical-molecular mechanisms has been possible after studies of phosphate-wasting bone diseases, such as hypophosphatemic rickets (2). Cumulated data from in vitro and in vivo researches, with human and animal models, converge toward the hypothesis of a bone-renal axis controlling this homeostasis (1,3).
Rickets are metabolic bone diseases, characterized by impaired mineralization of the osteoid matrix during growth, affecting epiphyseal growth plate and compromising both cortical and trabecular bone. Osteomalacia, at the other hand, refers to a deficient mineralization of the osteoid matrix in modeling and remodeling sites (4), injuring the corticoendosteal tissue. In pediatric ages, rickets and osteomalacia may happen simultaneously, but after growth plate closure only osteomalacia may occur. Rickets and osteomalacia can result from vitamin D deficiency (nutritional or metabolic disturbances), calcium and/or phosphorus deficiency and primary bone mineralization defects (hereditary hypophosphatasia) (5).
Rickets and osteomalacia secondary to vitamin D deficiency and/or low calcium intake in the XXI century has been a delicate theme. Recently, local and regional studies have recorded higher than expected vitamin D deficiency prevalence, mainly in high latitude regions, due to insufficient ultraviolet light exposure, but also in other countries, like France and United States, affecting urban healthy adolescents (6-8). This kind of rickets may also be related to specific situations, such as whole body covering dressing (due to religious habits), people with dark pigmented skins who live in temperate climate (due to melanin competition for ultraviolet rays), elderly people, vegetarians, restrictive diets and exclusive breastfeeding for long periods (9,10).
In developed countries, hypophosphatemic rickets (HR) are the main cause of inherited rickets (11). Initially, HR was seen as Vitamin D resistant rickets, once the lack of response to therapy was thought to be a Vitamin D resistant state. Since than, many studies have been performed and they managed to show that this disturb was consequence of a hyperphosphaturic disorder (12). Important advances in our current understanding of the molecular and biochemical bases of HR have been brought after the development of new molecular techniques and improved genomic databases (2).
HR comprises a set of disorders, inherited and acquired, that shares a common pathophysiology: diminished phosphate reabsorption in renal tubules (3,13-15). This process, established by an impaired phosphorus renal handling, leads to chronic hyperphosphaturia and hypophosphatemia, which are associated to unsuitable normal or low levels of 1,25(OH)2VitaminD3. This phosphorus-wasting disturbance affects bone metabolism, yielding rickets and osteomalacia in pediatric patients and osteomalacia in adults.
X-linked hypophosphatemic rickets (XHR), autosomic dominant hypophosphatemic rickets (ADHR) and tumor-induced osteomalacia (TIO) are the main syndromes that integrate the hypophosphatemic rickets, being the XHR the most frequent one (16,17).
Although these diseases have different etiopathogenies, biochemical and molecular evidences point to a common pathophysiological basis: increased activity of a phosphaturic factor (2,13-15). There are some other bone diseases associated with phosphaturic imbalance, such as hereditary hypophosphatemic rickets with hipercalcemia (HHRH) and McCune Albright Syndrome (3).
PHOSPHORUS METABOLISM
Phosphorus plasma levels are kept within a narrow range and they are under the influence of many factors, such as age, sex, diet, plasma pH and hormones. The appropriate phosphorus serum concentrations are essential to bone mineralization. Beyond this, phosphorus regulation is mandatory to the integrity of some biochemical process, like intracellular energy generation and transference, signaling transduction, nucleotides metabolism and some enzymatic activities (20).
Approximately 70% of plasma phosphorus is present in an organic form and 30% in an inorganic form. Just 15% of inorganic phosphorus is bound to proteins, while 85% circulates as free phosphate ions or in complex compounds with sodium, magnesium and calcium. Inorganic orthophosphate is the circulating compound used for measurement of phosphorus plasma concentrations. One relevant point is that phosphatemia decreases progressively with age. Reference ranges are different according to ages: before 3 months of age, normal values stay between 4.8 and 7.4 mg/dl; from 1 to 5 years, between 4.5 and 6.2 mg/dl; from 6 to 12 years, between 3.6 and 5.8 mg/dl and in young adults it ranges from 2.5 to 4.5 mg/dl (21).
Phosphorus metabolism is intricate. Phosphatemia levels result from the balance among phosphorus intestinal absorption, tubular reabsorption and ion exchange between intracellular and bone pools. While intestinal phosphorus absorption is adjusted rather slowly for maintaining or regaining the mineral homeostasis, renal phosphorus balance can be adjusted quite fast.
The main organ responsible for phosphorus homeostase is the kidney (2,3). In addition to the well-known PTH-calcitriol axis, other components participate actively on this regulation. Amongst the most important contributing data to the knowledge of phosphorus physiology, one can point out the elucidation of part of the role of sodium-dependent phosphate co-transporter proteins (NaPiT-I, II and III) and the fibroblast growth factor 23.
NaPiT co-transporter system is present in many tissues. NaPiT-IIb is found mainly in the small bowel (jejune), where it participates of the dietary phosphorus absorption process (22).
Renal proximal tubular reabsorption of phosphorus is a critical variable for the systemic phosphorus homeostasis. This process encompasses a secondary active phosphorus transport mechanism, in which sodium-phosphate (Na/Pi) co-transport systems play a primordial role. Type I (NaPiT-I) and types IIa and IIc (NaPiT-IIa e IIc) are expressed on the brush border membrane of proximal tubules, where 85% of the filtered inorganic phosphorus (Pi) is reabsorbed, resting just a small amount to be reabsorbed in the Henle loop and in distal tubules (23,24). In the proximal tubule, a Na+/K+ ATPase displaces sodium outward the cell. Due to a sodium concentration gradient and NaPiT-II co-transporters, sodium returns to the cell, bringing phosphorus together.
Type III co-transporters (NaPiT-III) are retroviral receptors expressed in many tissues, responsible for less than 1% of the mRNA related to NaPiT co-transporters and they probably act as house-keeping transporters (24).
FGF-23, the largest member of the fibroblast growth factor family (FGF), contains 251 amino acids and it is encoded by a gene in 12p13 (34). It is synthesized by osteogenic cells, osteoblasts and osteocytes. This protein displays NaPiT-IIa and IIc inhibition activity and exerts a regulatory role in phosphaturia (3,25), being postulated that it is a "phosphaturic factor" also called phosphatonin. FGF-23 also inhibits renal 25(OH)-1-a-hydroxylase activity, leading to a decreased calcitriol synthesis (3). The exact role of this factor in phosphorus homeostase is not completely known yet. Its plasma levels are positively correlated to dietary phosphorus and to phosphaturia and negatively related to 1,25(OH)2 Vitamin D3 plasma levels. Animal models show that 1,25(OH)2VitaminD3 and phosphatemia regulate, independently, FGF-23 concentrations (27).
The phosphorus tubular reabsorption is also stimulated by hormones and cytokines, such as PTH, GH, IGF-1, insulin, epidermal growth factor, thyroid hormones, glucocorticoids and 1,25(OH)2Vitamin D3 (2). Dietary phosphorus ingestion and its renal tubular reabsorption intensity are inversely related. PTH, calcitonin, natriuretic factor and glucocorticoids inhibit this reabsorption. PTH and FGF-23 act as phosphaturic agents, reducing phosphorus reabsorption in the proximal tubules, through NaPiT-II co-transporters inhibition. Although PTH main function is related to calcium homeostasis, it also takes part in phosphorus metabolism. This hormone inhibits NaPiT-II expression, promoting internalization of this co-transporter and its lisossomal degradation (28,29), reducing its synthesis (30), thus down-regulating its expression on the cell surface.
HYPOPHOSPHATEMIC RICKETS PATHOPHYSIOLOGY
X-linked hypophosphatemic rickets (XHR), autosomic dominant hypophosphatemic rickets (ADHR), tumor induced osteomalacia (TIO), fibrous dysplasia and McCune Albright Syndrome share a common underlying pathophysiological condition: increased phosphorus renal loss secondary to augmented FGF-23 plasma levels and activity.
In XHR, hyperphosphaturia derives from mutations in the PHEX gene (Phosphate regulating gene with homologies to Endopeptidases on the X-chromossome) (1-3,13-16). This gene, whose locus is Xp22.1, encodes a membrane endopeptidase (Zn-metaloprotease), called Phex, mainly expressed in bone and teeth. Recent data indicate that this enzyme integrates the phosphate-handling processes, regulating FGF-23 synthesis and/or degradation.
Some studies have shown that Phex promotes FGF-23 cleavage, originating two smaller and inactive peptides (31). Physiologically, Phex-induced FGF-23 cleavage would regulate phosphorus reabsorption in proximal tubules: an increase in Phex action would increase the inactivation of FGF-23 and increase phosphorus reabsorption.
The mutations in PHEX gene associated to XRH yield a decrease or absence of action of Phex, thus increasing FGF-23 actions and inhibiting the inactivation of NaPiT-II co-transporters, leading to increased phosphorus tubular reabsorption (1-3,13-16). Although Phex actions on FGF-23 would explain the pathophysiology of XHR, some studies have shown FGF-23 cleavage by convertases, not by Phex endopeptidase (32). Other researches did not manage to confirm that FGF-23 actually is the Phex direct substrate (33), suggesting that this metaloendopeptidase might act through another regulatory mechanism on FGF-23. Some authors point out the likelihood of being intermediate pathways between Phex and FGF-23, which would promote FGF-23 inactivation and that Phex would trigger chain reactions as well, in order to modulate osteoblast FGF-23 expression (33).
Then, it is postulated that in XHR PHEX gene mutations inhibit FGF-23 inactivation and/or promote increase of its plasma levels, yielding to hyperphosphaturia (2,3,34-36). In addition to that, FGF-23 increased action inhibits 1a-hydroxylase, the responsible enzyme for 25(OH)VitaminD (calcidiol) conversion to its active form, 1,25(OH)2VitaminD (calcitriol). Through this mechanism, the rise of calcitriol in the plasma is avoided, contributing to enhance hypophosphatemia (2). This way, rickets related to FGF-23 increased activity do not evolve with calcitriol higher levels, although hypophosphatemia would be a natural stimulus for that augmentation.
ADHR is less frequent genetic cause of hypophosphatemic rickets secondary to hyperphosphaturia. It is caused by activating missense mutations in FGF-23 gene, affecting one of the two arginines in locus 176 or 179. Identified mutations are R179Q, R176Q, R179W and they alter the natural sequence that is recognized by proteases, making them resistant to proteolysis (35,37). The outcome is an increased plasma level of FGF-23. The mutant FGF-23 bears higher biological potency than the wild type (38) in decreasing the expression of co-transporters NaPiT-II encoding genes and as inhibitor of 1a-hydroxylase activity (2,39).
TIO is a paraneoplastic syndrome that leads to an acquired hypophosphatemic hyperphosphaturic type of rickets. It is related to mesenchymal or mixed connective tissues tumors, generally benign, that occur chiefly in head and neck, and less frequently in bones and other tissues (40,41). The most common types are the vascular neoplasms (hemangiopericytomas). Other types include fibromas, condrosarcomas, hystiocytomas, neuroblastomas and prostate carcinoma (40,41).
These tumors overproduce FGF-23 and this excessive amount cannot be adequately degraded by Phex (35), taking into account the hypothesis that this factor is the peptidase substract, or by another specific proteolytic enzyme. This results in increased phosphaturia and inhibition of 1a-hydroxylase (2,40). Although FGF-23 levels are generally elevated, they may be inside normal range in some cases (36,41).
Two other proteins have been found to be over-expressed by this tumors and they also seem to be related to bone mineralization disturbances: MEPE (matrix extracelular phosphoglicoprotein) e sFRP-4 (secreted Frizzled Related Protein). Initially, it was thought that MEPE would suffer proteolysis by Phex. Nevertheless, this hypothesis was not confirmed by in vitro models. Some studies suggest that MEPE would exert its role on mineralization through not elucidated Phex-dependent mechanisms (3). The sFRP-4 protein is a renal Wnt signaling extracellular antagonist, which inhibits NaPiT-II co-transporter and promotes phosphaturia (42). Nevertheless, the mechanisms that these proteins regulate phosphorus homeostasis have not been elucidated yet.
The epidermal nevus syndrome and type 1 neurofibromatosis may, rarely, be associated with hypophosphatemic rickets and hyperphosphaturia due, probably, to increased secretion of FGF-23 by cells from nevus or neurofibromas (43,44). Although there are some variants of TIO, the resection of the nevus may not result in normalization of clinical and laboratory alterations (45).
The fibrous dysplasias, monostotic or polyostotic, are caused by activating mutations of GNAS1 gene that result in constitutive activating of adenyl cyclase by mutant a subunit. The involvement of cells of the osteogenic lineage affects the osteoprogenitor cells and differentiated osteoblasts, resulting in marrow infiltration and fibrosis, reduction of hematopoietic tissue and formation of abnormal bone tissue with altered trabeculae, collagen orientation and biochemical composition (46). The polyostotic fibrous dysplasia may occur alone or associated to other signs and symptoms, as in McCune-Albright syndrome which, beyond the bone lesions, is characterized by large and hyperpigmented lesions of skin, with irregular borders and a typical brown coloration ("café-au-lait") and hyperfunction of peptide hormones that act through a coupled G receptor (gonadotrophins, TSH, PTH, ACTH and GHRH), where the gonadotrophin independent precocious puberty is the most common hormone dysfunction. In fibrous dysplasia, and especially in polyostotic lesions (associated or not to McCune-Albright syndrome), the excessive production of FGF-23 by osteoprogenitor cells may result in hyperphosphaturia, reduction of the activity of 1a-hydroxylase and hypophosphatemic rickets or osteomalacia (26,47).
The hereditary hypophosphatemic rickets with hypercalciuria (HHRH) is an autosomal recessive disease characterized by hypophosphatemia, hyperphosphaturia, normal PTH serum levels and increased plasma levels of 1,25(OH)2 Vitamin D3, that is responsible for hypercalciuria (2,18). The increased plasma levels of 1,25(OH)2VitaminD3 and the hypercalciuria distinguish the HHRH from the other causes of hypophosphatemic rickets. The most recent evidences show that HHRH is associated to mutations that affect the two alleles of gene SLC34A3, responsible for NaPiT-II co-transporter activity, rendering the co-transporter inactive (19). The heterozygotes for mutation in SCL34A3 gene present hypercalciuria with mild or absent hypophosphatemia (19).
Studies of the 3 main types of hypophosphatemic rickets (XHR, ADHR and TIO) suggest a common pathophysiologic underlying mechanism: hyperphosphaturia secondary to reduction of tubular phosphate reabsorption. Researches on these diseases indicate the existence of a metabolic bone-renal axis capable to regulate the phosphorus homeostasis and the bone mineralization. FGF-23 seems to have a central role on this axis (2,3,34) through its phosphaturic action and its autocrine action on osteoblasts, modulating, thus, bone mineralization. Alterations in FGF-23 metabolism, due to inhibition of its proteolytic cleavage or to its increased resistance to proteolysis, trigger disturbances in phosphorus homeostasis and in bone-mineral metabolism.
Some authors suggest that the osteoblasts are the candidate cells to assume the metabolic coordination of this bone-renal axis, once they synthesize proteins that have important role in phosphorus homeostasis and in osteoid mineralization, including FGF-23, Phex, MEPE, and express elements that regulate bone mineralization, bone mass and renal phosphate conservation (3).
CLASSIFICATION
The growing importance of FGF-23 controlling phosphorus homeostasis and phosphaturia has brought the interest to propose the following classification for hypophosphatemic rickets or osteomalacia.
Causes of hypophosphatemic rickets or osteomalacia with increased plasma FGF-23
· Increased plasma FGF-23 due to inadequate proteolysis:
- Impaired FGF-23 proteolysis caused by deficient action of mutant Phex: XHR
- Inadequate proteolysis of FGF-23 due to proteolysis-resistant mutant FGF-23: ADHR
· Increased production of FGF-23:
- Increased production of FGF-23 by mesenchymal tumors: TIO
- Increased production of FGF-23 by osteoprogenitor cells and osteoblasts: fibrous dysplasia, McCune-Albright syndrome
- Increased production of FGF-23 by other causes: epidermal nevus syndrome, type 1 neurofibromatosis
Causes of hypophosphatemic rickets or osteomalacia with normal or decreased plasma FGF-23:
· HHRH
· Fanconi's renal syndrome
CLINICAL PICTURE
In infants and children the main clinical manifestations of hypophosphatemic rickets are similar to those observed in nutritional rickets, except for some particularities that will be discussed. The most characteristic findings include: growth retardation, deformities in upper and lower limbs (genu varum or genu valgum) generally noted when the infant begins to creep or to walk, metaphyseal widening, palpable enlargement of the costochondral junctions (rachitic rosary), frontal prominence, horizontal depression along the lower border of the chest (Harrison's groove), insufficient weight gain. The thickness of central parts of parietal and frontal bones may alter the shape of the head. Recurrent fractures, although not common, may worsen bone deformities. Figures 1 and 2 show some of these characteristics, in 2 patients with hypophosphatemic rickets followed at the Pediatric Endocrinology Unit of Instituto da Criança (HCFMUSP).
X-linked hypophosphatemic rickets
The XHR is the main cause of rickets in developed countries, with an incidence of 1 case in each 20,000 persons. The disease affects the hemizygous boys and heterozygous girls. There is no correlation between the severity of the disease and the type or localization of PHEX gene mutations (48). The severity of the disease is variable even among affected individuals from the same family. The mild forms may present just with hypophosphatemia in the absence of bone lesions (17). Birth length is normal, but a reduced growth velocity in the first years of life is responsible for short stature during the childhood. The lower limbs are more severely affected than the upper limbs. Final height compromising is due to the reduced growth before the diagnosis (49). Differently from vitamin D dependent rickets or from other causes of hypophosphatemic rickets (ADHR, TIO and HHRH), in XHR muscle weakness and hypotonia are absent (50). XHR is also characterized by dental damages due to dentin under-mineralization and tendency to teeth fall and dental abscesses (51). These dental alterations are more severe in adolescents and in male adults (48).
Other complications observed in adults with X-linked osteomalacia include enthesopathy (ossification of tendon, ligament and articular capsule), pseudo-fractures, articular pain, osteoarthritis (mainly affecting lower limbs) and neurosensorial hypoacusia (52).
Autosomal dominant hypophosphatemic rickets
Differently from other hereditary causes of hypophosphatemic rickets, in ADHR the age of manifestation is quite variable and the disease may appear only in adolescence or during adulthood (53). When manifested after the puberty, the most common characteristics of ADHR are bone pain, pseudo-fractures and muscle weakness (2,53). The absence of short stature and lower limb deformities in some of these patients suggest a not so intense hypophosphatemia during infancy and childhood (53). Dental abscesses may occur. A recent study describes the variability in clinical manifestations in one family, including asymptomatic adults, others with bone pain in the sixth decade of life and children with symptoms since the first years of life (54). Another interesting aspect of this rickets is the likelihood of spontaneous decreasing in hyperphosphaturia (53). It has not yet been clarified how and why the genetic defects manifest only after the first years of life and the mechanisms implicated in spontaneous resolution of hyperphosphaturia.
Tumors-induced osteomalacia
TIO is a rare paraneoplasic syndrome where the clinical-radiological signs are similar to those observed in other causes of rickets and osteomalacia (40). However, bone pain and fractures are usually more severe than in XHR and muscle weakness is frequent (40). It is interesting to note that the bone mineral disturbances are the most important manifestations of these tumors. Although TIO may affect children, generally it occurs in older ages compared to XHR. The tumors associated to TIO are, generally, very small, rather benign than invasive and hardly diagnosed by routine physical examination or radiographs. The paranasal sinuses, neck and mandible are common sites of these tumors and they must be accessed by computed tomography or nuclear magnetic resonance imaging (40).
Hereditary hypophosphatemic rickets with hypercalciuria
In patients with HHRH the classical signs of rickets are accompanied by muscle weakness and bone pain, but dental alterations are not present (2).
DIAGNOSIS
The diagnosis of hypophosphatemic rickets is based in the clinical picture and laboratory and radiograph alterations.
Laboratory findings
XHR, ADHR and TIO are all characterized by hypophosphatemia, hyperphosphaturia, increased plasma level of alkaline phosphatase, normocalcemia and normal or reduced calciuria. It is important to remember that phosphate plasma levels vary according to age (21).
Phosphaturia may be evaluated through phosphorus tubular reabsorption (PTR):

Pu and Pp, Cr.u and Cr.p mean urine and plasma phosphorus concentration and urine and plasma creatinin concentration, respectively (all variables must be expressed in the same unit).
Hyperphosphaturia is characterized by PTR values inferior to 85%, concomitant to hypophosphatemia (50).
Alkaline phosphatase plasma evaluation is important for rickets diagnosis and as a parameter for disease control and therapy efficacy as well. Adequately treated patients evolve with a progressive reduction in plasma alkaline phosphatase.
In XHR, ADHR, TIO and in other causes of hypophosphatemic rickets due to increased activity of FGF-23, plasma calcitriol is inappropriately normal or reduced, secondary to inhibition of 1a-hydroxylase (2,34,40,50). These levels of calcitriol justify the normal or upper normal PTH plasma levels found in these hypophosphatemic rickets (5,50).
The diagnosis of XHR in the first months of life is difficult, even when there are familial cases. Usually, infants with familial antecedents for XHR can be diagnosed from the 6th month of life, based on increased alkaline phosphatase and reduced renal phosphate reabsorption (55).
Biochemically, HHHR differs from XHR, ADHR, and TIO in hypercalciuria, elevated calcitriol concentration and low serum PTH (2). This aspect gains importance in terms of therapy.
Imaging studies
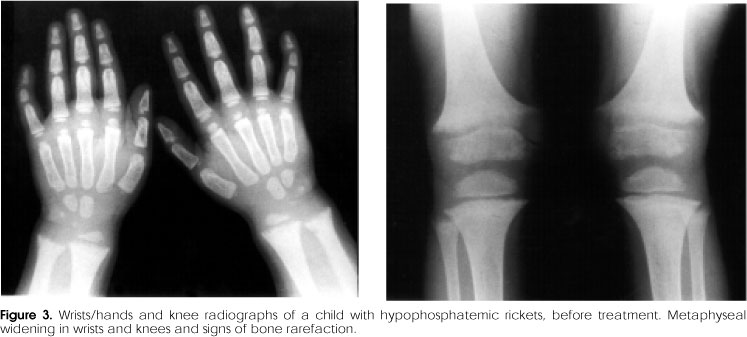
In XHR, the radiologic alterations are seen in tibia, distal femur, radio and ulna, and reflect loss of definition, widening, and calyx image in the provisional calcification zone in the growth plate (17) (figure 3). These findings are less severe than those seen in the vitamin D-deficient rickets and the signs are more intense in lower limbs (50).
Once TIO is suspected, a careful imaging study has to be done, through cranial CT and MRI of facial sinuses and mandible. To localize the tumor on bone, pentreotide or 111I octreotide scintigraphy is mandatory (40).
When bone fibrous dysplasia is suspected, 99Tc cintigraphy is useful.
During investigation of rickets occurring in older children and adolescents, especially in the absence of familial history, it is very important to consider other differential diagnoses leading to hyophosphatemic hyperphosphaturic rickets, such as ADHR, TIO, bone fibrous dysplasia, and renal Fanconi syndrome. Certain clinical features help in differentiating these conditions.
Fanconi syndrome differs from XHR because the former is characterized by tubular renal acidosis, with metabolic acidosis and renal tubular dysfunction, leading to decreased phosphate reabsorption, increased bicarbonate renal wasting, glucosuria, and aminoaciduria (56).
FGF-23 dosage
So far, plasma dosage of FGF-23 has had scientific purposes, characterizing diseases that cause hypophosphatemic rickets and hyperphosphaturia. However, it is possible that, in the future, this dosage becomes one more lab resource to diagnose and follow-up such diseases. Lab assays that detect only the whole FGF-23 molecule seem to be more trustable than the methods that evaluate the terminal carboxi end (57).
TREATMENT
Clinical treatment of hypophosphatemic rickets aims at minimizing the metabolic disturbances, reducing bone deformities and improving growth velocity, although the hyperphosphaturia persists, once the treatment does not alter the impaired tubular phosphate reabsorption.
In patients with TIO, the basic management is surgical removal of the tumor, which allows correction of the excess of phosphaturic factor and final cure of the condition. However, not all patients show good clinical response to therapy and these results reflect not only the patient compliance, but also the age at the beginning of therapy (49).
The treatment of children and adolescents with XHR includes phosphate administration (neutral salts of inorganic sodium and potassium phosphate) and calcitriol. Elementary phosphorus is given at a dose of 3060 mg/kg/day, divided in 46 fractions (5). Start with 30 mg/kg/day increasing gradually according to the biochemical profile of the patient. It is important not to administer phosphate with milk ingestion, since the calcium contained in these dairy products interferes with phosphate intestinal absorption.
Phosphorus absorption is slower in capsule or tablet presentation when compared to liquid formulations. When possible, it is better to use capsules or pills (58). Transient side effects can follow phosphate ingestion, such as abdominal pain and osmotic diarrhea (58). Calcitriol is used in the dose of 3070 ng/kg/day (5,58), qd or bid. The treatment of XHR has to be kept till the end of statural growth (58).
With adequate treatment, the serum levels of alkaline phosphatase (AlP) decrease, reaching normal or slightly elevated levels. The dose of phosphorus has to be increased if the AlP levels increase (49). The fall in AlP is more intense when treatment is started in the first months of life, compared to treatments started after two years of age. In these patients, AlP may never normalize (49).
In patients with XHR, it is recommended withdrawing the medication one week before orthopedic surgeries, aiming at avoiding hypercalcemia caused by bone reabsorption due to prolonged immobilization. The therapy should be restarted as soon as the patient gains conditions to walk (50).
Early treatment of XHR can minimize growth impairment, limb deformities, and teeth anomalies (49,51,60). However, the growth deficit accumulated in the first years of life is usually permanent, especially when treatment has begun at a later age. As XHR children are born with a normal length and have a normal growth spurt, it is believed that the final short stature is, at least in part, due to loss of height in the first pre-treatment years (40).
Many studies have evaluated the role of recombinant human growth hormone (rhGH) in the treatment of children with XHR. The results have varied in terms of reestablishment of phosphorus homeostasis and the achievement of a better growth performance (62-65). In two studies improvement in growth was noticed but with increased disproportion between upper and lower segments, predominating the trunk growth (66,67). Systematic review on the use of rhGH in patients with XHR does not show conclusive evidence in favor of rhGH changing the statural growth, mineral metabolism, mineral density nor body proportions (68).
The treatment of ADHR and other causes of hypophosphatemic rickets with hyperphosphaturia, due to increased action of FGF-23 must follow the same principles mentioned in the treatment of XHR.
In cases of TIO, the excision of the tumor to correct the biochemical alterations is the key procedure (40,69) since it suppresses the source of systemic phosphaturic factors. The serum levels of phosphate and calcitriol as well as the tubular phosphate reabsorption become normal in hours or days, while the bone remodeling markers (calcitonin and alkaline phosphatase) and skeletal deformities take more time to be corrected (40).
With regard to HHRH, its treatment implies only elementary phosphorus administration, with reversal of the biochemical and bone alterations. Calcitriol is not indicated, due to hypercalciuria and to the high calcitriol levels characteristic of the disease.
COMPLICATIONS
The main complications of XHR are related to the treatment itself, since it is not easy to find a balance between phosphate and calcitriol doses that are able to solve the clinical picture without yielding hyperparathyroidism or hypercalciuria. Among them, we include secondary and tertiary hyperparathyroidism and nephrocalcinosis.
Doses of phosphate above 50 mg/kg/day can induce PTH stimulation and give rise to secondary hyperparathyroidism with reduction of calcemia through the direct action of phosphate over the PTH secretion (59,70). The persistency of hyperparathyroidism for long periods of time can lead to autonomous function of parathyroid, characterizing the tertiary hyperparathyroidism, which, although rare, is severe, leading to intense bone resorption, nephrocalcinosis, and renal insufficiency. Tertiary hyperparathyroidism can lead to hypercalcemia, while in the secondary form, calcium levels are normal or decreased (70). In the treatment of XHR the following factors favor the progression to tertiary hyperparathyroidism: early start of treatment, longer duration of treatment, high doses of elementary phosphorus (100 mg/kg/day), and very high PTH plasma levels (around 400 pg/mL) (70).
XHR patients under treatment can present interstitial nephrocalcinosis, due to deposition of calcium phosphate in the renal pyramids, which must be followed-up with ultra-sound exams. Nephrocalcinosis is not directly related to worsening of renal function (58). These patients can also present vascular disturbances, especially systemic hypertension, not related to nephrocalcinosis but to the secondary or tertiary hyperparathyroidism (71).
In children and adolescents with XHR mineral bone density can be reduced in the appendicular skeleton and increased in the lumbar spine (72). This pattern tends to be kept in the adult age and does not predispose to osteoporotic fractures (73).
MONITORING THE TREATMENT
It has been recommended that XHR patients under treatment be evaluated every three months (5). Lab work-up has to be made monthly at the beginning and every three months later on, and includes serum dosage of calcium, phosphorus, creatinine, alkaline phosphatase as well as urinary dosage of calcium and creatinine in 24h-urine sample. Serum PTH has to be evaluated every six months, and kidney ultra-sound every 6 to 12 months (17).
The calcitriol dose must be adjusted according to the serum levels of PTH and the urinary calcium concentration. PTH above the upper normal limit implies increase in the calcitriol dose and/or decrease of phosphate dose. The main effect of excessive dose of calcitriol is hypercalciuria, defined as urinary excretion of calcium above 4 mg/kg/day or calcium/creatinine ratio above 0.7 in the first year of life or above 0.3 after the first year (59). If hypercalciuria is present, reduce the calcitriol dose.
Concerning the other causes of hypophosphatemic rickets, we should follow the same principles outlined before, based on the etiopathogenic characteristics of each syndrome.
CONCLUSION
The complex interaction among the components of the metabolic axis implied in the phosphorus homeostasis and bone mineralization has been better understood in the last years.
This knowledge is due, partially, to a better understanding of pathophysiology of hypophosphatemic disturbances and its implications in the bone-mineral metabolism. However, many steps of this process and the mechanisms of communication among the actors in this delicate system have not yet been clarified.
Accumulated evidence both in in vivo as in vitro studies, in animal and human models have pointed to the theory of a bone-renal axis which promotes and regulates homeostasis between phosphorus and bone tissue mineralization. The physiology of the osteoblast allows us to infer that this cell has the basic equipment to coordinate such a process. All these data are strategic and precious information to open up novel perspectives for new, more efficient and with less side effects therapeutic options to treat the various forms of hypophosphatemic rickets.
Received in 04/01/06
Accepted in 05/08/06
- 1. Rowe P. The wrickkened pathways of FGF-23, MEPE and PHEX. Crit Rev Oral Biol Med 2004;15(5):264-81.
- 2. de Beur SMJ, Levine MA. Molecular pathogenesis of hypophosphatemic rickets. J Clin Endocrinol Metab 2002;87:2467-73.
- 3. Quarles LD. Evidence for a bone-kidney axis regulating phosphate homeostasis. J Clin Invest 2003;112:642-6.
- 4. Wharton B, Bishop N. Rickets. Lancet 2003;362:1389-400.
- 5. Root AW, Diamond FB Jr. Disorders of calcium metabolism in the child and adolescent. In: Sperling MA (ed). Pediatric Endocrinology 2nd ed. Philadelphia: Saunders, 2002 p. 646.
- 6. Holick MF. Vitamin D a millenium perspective. J Cel Biochemist 2003;88:296-307.
- 7. Garabédian M, Menn S, Walrant-Debray O, Teinturier C, Delaveyne R, Roden A. Prévention de la carence en vitamine D chez l'enfant et l'adolescent. II. Validation d'un abaque décisionnel non invasif prenant en compte l'exposition solaire et les apports exogènes de vitamine D. Archives de Pediatrie 2005; 12(4):410-9.
- 8. Gordon C, DePeter K, Feldman H, Grace E, Jean Emans S. Prevalence of Vitamin D deficiency among healthy adolescents. Arch Pediatr Adolesc Med 2004;158:531-7.
- 9. Mughal Z. Rickets in childhood. Semin Musculoskelet Radiol 2002;6:183-90.
- 10. Pettifor JM. Nutritional and drug-induced rickets and osteomalacia. In: Favus MJ (ed). Primer on the metabolic bone diseases and disorders of mineral metabolism 5th ed. Washington: American Society for Bone and Mineral Research, 2003 p. 399.
- 11. Garg RK, Tandon N. Hypophosphatemic rickets: easy to diagnose, difficult to treat. Indian J Pediatr 1999;66:849-57.
- 12. Winters R, Grahan J, Williams T, McFalls V, Burnett C. A genetic study of familial hypophosphatemia and vitamin D resistant rickets with a review of the literature. Medicine 1958;37:97-142.
- 13. Rowe OS. The molecular background to hypophosphatemic rickets. Arch Dis Child 2000;83:192-4.
- 14. Cho HY, Lee BJ, Kang JH, Ha IS, Cheong HI, Choi Y. A clinical and molecular genetic study of hypophosphatemic rickets in children. Pediatr Res 2005;58(2): 329-33.
- 15. Bielesz B, Klaushofer K, Oberbauer R. Renal phosphate loss in hereditary and acquired disorders of bone mineralization. Bone 2004;35:1229-39.
- 16. Saggese G, Baroncelli GI. Hypophosphatemic rickets. Horm Res 2000;53(suppl. 3):57-60.
- 17. Tenenhouse HS, Econs MJ. Mendelian hypophosphatemias. In: Scriver CR (ed). The Metabolic and Molecular Bases of Inherited Disease 8th ed. McGraw-Hill, 2001 pp. 5039-67.
- 18. Tieder M, Arie R, Bab I, Maor J, Liberman UA. A new kindred with hereditary hypophosphatemic rickets with hypercalciuria: implications for correct diagnosis and treatment. Nephron 1992;62(2):176-81.
- 19. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet 2006;78(2):179-92.
- 20. Berndt TJ, Schiavi S, Kumar R. "Phosphatonins" and the regulation of phosphorus homeostasis. Am J Physiol Renal Physiol 2005;289(6):F1170-82.
- 21. Portale AA. Blood calcium, phosphorus, and magnesium. In: Favus MJ (ed). Primer on the metabolic bone diseases and disorders of mineral metabolism 5th ed. Washington: American Society for Bone and Mineral Research, 2003 pp. 151-4.
- 22. Hilfiker H, Kvietikova II, Hartmann CM, Stange G, Murer H. Characterization of the human type II Na/Pi-cotransporter promoter. Pflugers Arch 1998;436:591-8.
- 23. Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, et al. Growth-related renal type II Na/Pi cotransporter. J Biol Chem 2002;277:19665-72.
- 24. Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflugers Arch 2004;447:763-7.
- 25. Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 2003;112:683-92.
- 26. Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 2005;90:1519-24.
- 27. Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 2005;280:2543-9.
- 28. Murer H, Biber J. Molecular mechanisms of renal apical Na/phosphate cotransport. Ann Rev Physioll 1996;58:607-18.
- 29. Pfister MF, Ruf I, Stange G, Ziegler U, Lederer E, Biber J, et al. Parathyroid hormone leads to the lysomal degradation of the renal type II Na/Pi cotransporter. Proc Natl Acad Sci USA 1998;95:1909-14.
- 30. Malmström K, Murer H. Parathyroid hormone regulates phosphate transport in OK cells via an irreversible inactivation of a membrane protein. FEBS Lett 1987;216:257-60.
- 31. Bowe AE, Finnegan R, de Beur SMJ, Cho J, Levine MA, Kumar R, et al. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun 2001;284:977-81.
- 32. Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone 2004;35:455-62.
- 33. Liu S, Guo R, Simpson LG, Xiao Z-S, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem 2003;278(39):37419-26.
- 34. Strewler GJ. FGF23, hypophosphatemia, and rickets: has phosphatonin been found? Proc Natl Acad Sci USA 2001;98:5945-6.
- 35. Quarles LD, Drezner MK. Pathophysiology of X-linked hypophosphatemia, tumor-induced osteomalacia, and autosomal dominant hypophosphatemia: a perPHEXing problem. J Clin Endocrinol Metab 2001;86: 494-6.
- 36. Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 2003;348:1656-63.
- 37. White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int 2001;60:2079-86.
- 38. Bai XY, Miao D, Goltzman D, Karaplis AC. The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem 2003;278:9843-9.
- 39. Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitaminD3 production. J Biol Chem 2003;278:2206-11.
- 40. Carpenter TO. Oncogenic osteomalacia a complex dance of factors. N Engl J Med 2003;348:1705-8.
- 41. Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM, Shulman D, et al. Sensitivity of fibroblast growth factor 23 measurements in tumor induced osteomalacia. J Clin Endocrinol Metab 2006;91(6): 2055-61.
- 42. Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest 2003;112(5):642-6.
- 43. Konishi K, Nakamura M, Yamakawa H, Suzuki H, Saruta T, Hanaoka H, et al. Hypophosphatemic osteomalacia in von Recklinghausen neurofibromatosis. Am J Med Sci 1991;301:322-8.
- 44. Hoffman WH, Jueppner HW, DeYoung BR, O'Dorisio MS, Given KS. Elevated fibroblast growth factor-23 in hypophosphatemic linear nevus sebaceous syndrome. Am J Med Genetics 2005;134A:233-6.
- 45. Kishida ES, Silva MAM, Pereira FC, Sanches Jr JA, Sotto MN. Epidermal nevus syndrome associated with adnexal tumors, spitz nevus, and hypophosphatemic vitamin D-resistant rickets. Pediatr Dermatol 2005;22:48-54.
- 46. Collins MT, Bianco P. Fibrous dysplasia. In: Favus MJ (ed). Primer on the metabolic bone diseases and disorders of mineral metabolism 5th ed. Washington: American Society for Bone and Mineral Research, 2003 pp. 466-70.
- 47. Yamamoto T, Imanishi Y, Kinoshita E, Nakagomi Y, Shimizu N, Miyauchi A, et al. The role of fibroblast growth factor 23 for hypophosphatemia and abnormal regulation of vitamin D metabolism in patients with McCune-Albright syndrome. J Bone Miner Metab 2005; 23:231-7.
- 48. Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, Cowell CT, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 2001;86:3889-99.
- 49. Mäkitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 2003;88:3591-7.
- 50. Mughal Z. Rickets in childhood. Semin Musculoskelet Radiol 2002;6:183-90.
- 51. Pereira CM, Andrade CR, Vargas PA, Coletta RD, Almeida OP, Lopes MA. Dental alterations associated with X-linked hypophosphatemic rickets. J Endodontics 2004;30:241-5.
- 52. Fishman G, Miller-Hansen D, Jacobsen C, Singhal VK, Alon US. Hearing impairment in familial X-linked hypophosphatemic rickets. Eur J Pediatr 2004;163:622-3.
- 53. Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: Clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab 1997;82(2):674-81.
- 54. Negri AL, Negrotti T, Alonso G, Pasqualini T. Distintas formas de presentación clínica de un raquitismo hipofosfatemico autosomico dominante por mutación del factor de crecimiento fibroblastico en una familia. Medicina (Buenos Aires) 2004;64:103-6.
- 55. Moncrieff MW. Early biochemical findings in familial hypophosphatemic, hyperphosphaturic rickets and response to treatment. Arch Dis Child 1982;57:70-2.
- 56. Sayer JA, Pearce SH. Diagnosis and clinical biochemistry of inherited tubulopathies. Ann Clin Biochem 2001; 38:459-70.
- 57. Ito N, Fukumoto S, Takeuchi Y, Yasuda T, Hasegawa Y, Takemoto F, et al. Comparison of two assays for fibroblast growth factor (FGF)-23. J Bone Miner Metab 2005;23:435-40.
- 58. Glorieux FH. Hypophosphatemic vitamin D-resistant rickets. In: Favus MJ (ed). Primer on the metabolic bone diseases and disorders of mineral metabolism 5th ed. Washington: American Society for Bone and Mineral Research, 2003 pp. 414-7.
- 59. Kruse K, Hinkel GK, Griefahn B. Calcium metabolism and growth during early treatment of children with X-linked hypophosphataemic rickets. Eur J Pediatr 1998;157:894-900.
- 60. Chaussain-Miller C, Sinding C, Wolikow M, Lasfargues JJ, Godeau G, Garabédian M. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: prevention by early treatment with 1-hydroxyvitamin D. J Pediatr 2003;142:324-31.
- 61. Mäkitie O, Doria A, Daneman A, Sochett E. Pubertal growth is normal in treated girls with X-linked hypophosphatemic rickets. J Bone Miner Res 2002; 17:S282
- 62. Seikaly MG, Brown R, Baum M. The effect of recombinant human growth hormone in children with X-linked hypophosphatemia. Pediatrics 1997;100:879-84.
- 63. Cameron FJ, Sochett EB, Daneman A, Kooh SW. A trial of growth hormone therapy in well-controlled hypophosphatemic rickets. Clin Endocrinol 1999;50:577-82.
- 64. Darendeliler F, Bas F, Karaaslan N, Hekim N, Budak R, Saka N, et al. The effect of growth hormone treatment on biochemical indices in hypophosphatemic rickets. Horm Res 2001;55:191-5.
- 65. Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G. Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr 2001;138:236-43.
- 66. Reusz GS, Miltényi G, Stubnya G, Szabó A, Horváth C, Byrd DJ, et al. X-linked hypophosphatemia: effects of treatment with recombinant human growth hormone. Pediatr Nephrol 1997;11:573-7.
- 67. Haffner D, Nissel R, Wühl E, Mehls O. Effects of growth hormone treatment on body proportions and final height among small children with X-linked hypophosphatemic rickets. Pediatrics 2004;113:e593-6.
- 68. Huiming Y, Meng M, Chaomin W, Fan Y. Recombinant growth hormone therapy for X-linked hypophosphatemia in children (Cochrane Review). In: The Cochrane Library, Issue 1, 2006 Oxford: Update Software.
- 69. Ward LM, Rauch F, White KE, Filler G, Matzinger MA, Letts M, et al. Resolution of severe, adolescent-onset hypophosphatemic rickets following resection of an FGF-23-producing tumour of the distal ulna. Bone 2004;34:905-11.
- 70. Mäkitie O, Kooh SW, Sochett E. Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets. Clin Endocrinol 2003;58:163-8.
- 71. Alon US, Monzavi R, Lilien M, Rasoulpour M, Geffner ME, Yadin O. Hypertension in hypophosphatemic rickets role of secondary hyperparathyroidism. Pediatr Nephrol 2003;18:155-8.
- 72. Shore RM, Langman CB, Poznanski AK. Lumbar and radial bone mineral density in children and adolescents with X-linked hypophosphatemia: evaluation with dual X-ray absorptiometry. Skeletal Radiol 2000;29:90-3.
- 73. Reid IR, Murphy WA, Hardy DC, Teitelbaum SL, Bergfeld MA, Whyte MP. X-linked hypophosphatemia: skeletal mass in adults assessed by histomorphometry, computed tomography, and absorptiometry. Am J Med 1991;90:63-9.
Publication Dates
-
Publication in this collection
14 Nov 2006 -
Date of issue
Aug 2006
History
-
Accepted
08 May 2006 -
Received
01 Apr 2006