Abstract
The aim of the present study was to determine the ventilation/perfusion ratio that contributes to hypoxemia in pulmonary embolism by analyzing blood gases and volumetric capnography in a model of experimental acute pulmonary embolism. Pulmonary embolization with autologous blood clots was induced in seven pigs weighing 24.00 ± 0.6 kg, anesthetized and mechanically ventilated. Significant changes occurred from baseline to 20 min after embolization, such as reduction in oxygen partial pressures in arterial blood (from 87.71 ± 8.64 to 39.14 ± 6.77 mmHg) and alveolar air (from 92.97 ± 2.14 to 63.91 ± 8.27 mmHg). The effective alveolar ventilation exhibited a significant reduction (from 199.62 ± 42.01 to 84.34 ± 44.13) consistent with the fall in alveolar gas volume that effectively participated in gas exchange. The relation between the alveolar ventilation that effectively participated in gas exchange and cardiac output (V Aeff/Q ratio) also presented a significant reduction after embolization (from 0.96 ± 0.34 to 0.33 ± 0.17 fraction). The carbon dioxide partial pressure increased significantly in arterial blood (from 37.51 ± 1.71 to 60.76 ± 6.62 mmHg), but decreased significantly in exhaled air at the end of the respiratory cycle (from 35.57 ± 1.22 to 23.15 ± 8.24 mmHg). Exhaled air at the end of the respiratory cycle returned to baseline values 40 min after embolism. The arterial to alveolar carbon dioxide gradient increased significantly (from 1.94 ± 1.36 to 37.61 ± 12.79 mmHg), as also did the calculated alveolar (from 56.38 ± 22.47 to 178.09 ± 37.46 mL) and physiological (from 0.37 ± 0.05 to 0.75 ± 0.10 fraction) dead spaces. Based on our data, we conclude that the severe arterial hypoxemia observed in this experimental model may be attributed to the reduction of the V Aeff/Q ratio. We were also able to demonstrate that V Aeff/Q progressively improves after embolization, a fact attributed to the alveolar ventilation redistribution induced by hypocapnic bronchoconstriction.
Effective alveolar ventilation; Mismatching ventilation/ perfusion; Pulmonary gas exchange
Braz J Med Biol Res, September 2006, Volume 39(9) 1197-1204
Mechanisms underlying gas exchange alterations in an experimental model of pulmonary embolism
 Correspondence and Footnotes
Correspondence and Footnotes
 J.H.T. Ferreira, R.G.G. Terzi, I.A. Paschoal, W.A. Silva, A.C. Moraes and M.M. Moreira
J.H.T. Ferreira, R.G.G. Terzi, I.A. Paschoal, W.A. Silva, A.C. Moraes and M.M. Moreira
Departamento de Cirurgia, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas, SP, Brasil
 Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
The aim of the present study was to determine the ventilation/perfusion ratio that contributes to hypoxemia in pulmonary embolism by analyzing blood gases and volumetric capnography in a model of experimental acute pulmonary embolism. Pulmonary embolization with autologous blood clots was induced in seven pigs weighing 24.00 ± 0.6 kg, anesthetized and mechanically ventilated. Significant changes occurred from baseline to 20 min after embolization, such as reduction in oxygen partial pressures in arterial blood (from 87.71 ± 8.64 to 39.14 ± 6.77 mmHg) and alveolar air (from 92.97 ± 2.14 to 63.91 ± 8.27 mmHg). The effective alveolar ventilation exhibited a significant reduction (from 199.62 ± 42.01 to 84.34 ± 44.13 mL) consistent with the fall in alveolar gas volume that effectively participated in gas exchange. The relation between the alveolar ventilation that effectively participated in gas exchange and cardiac output (VAeff/Q ratio) also presented a significant reduction after embolization (from 0.96 ± 0.34 to 0.33 ± 0.17 fraction). The carbon dioxide partial pressure increased significantly in arterial blood (from 37.51 ± 1.71 to 60.76 ± 6.62 mmHg), but decreased significantly in exhaled air at the end of the respiratory cycle (from 35.57 ± 1.22 to 23.15 ± 8.24 mmHg). Exhaled air at the end of the respiratory cycle returned to baseline values 40 min after embolism. The arterial to alveolar carbon dioxide gradient increased significantly (from 1.94 ± 1.36 to 37.61 ± 12.79 mmHg), as also did the calculated alveolar (from 56.38 ± 22.47 to 178.09 ± 37.46 mL) and physiological (from 0.37 ± 0.05 to 0.75 ± 0.10 fraction) dead spaces. Based on our data, we conclude that the severe arterial hypoxemia observed in this experimental model may be attributed to the reduction of the VAeff/Q ratio. We were also able to demonstrate that VAeff/Q progressively improves after embolization, a fact attributed to the alveolar ventilation redistribution induced by hypocapnic bronchoconstriction.
Key words: Effective alveolar ventilation, Mismatching ventilation/ perfusion, Pulmonary gas exchange
Introduction
Hypoxemia is one of the most important clinical signs of pulmonary embolism (PE). The complex abnormalities in gas exchange that occur are multiple, but the literature remains unassertive to elucidate the underlying mechanisms (1-10).
Analysis of the mechanisms involved in the genesis of this severe hypoxemia in humans with PE is difficult because of the multiple variables associated, including cardiopulmonary disease (8,9). Dantzker and Bower (8) stated that this difficulty exists even in experimental situations because of differences in species, the specific experimental circumstances, as well as the size, composition and quantity of the embolized material utilized, which may influence results in each individual model (11).
Several mechanisms are believed to be responsible for hypoxemia. These include low-partial oxygen pressure in mixed venous blood (PvO2), the mismatch between ventilation and perfusion, the intrapulmonary shunt, the diffusion limitation, and the severity of the embolism, impairing the identification of the real cause of hypoxemia (8,9). Recently, the mechanisms responsible for gas exchange have been investigated with the use of the multiple inert gas elimination technique (9).
In normal lungs there is equilibrium between alveolar ventilation and capillary perfusion at the proportion of approximately 1:1. The transfer of O2 is jeopardized when this equilibrium is altered and this proportion is reduced (2,4).
Pulmonary shunt occurs in humans and animals after PE, although its etiology has not been clearly established (8). Kasinski et al. (7) did not find a significant shunt in the embolization of the pulmonary circulation of dogs with autologous clots. Similarly, Wilson et al. (12) did not detect a shunt in clinical studies.
Mismatch between ventilation and perfusion has been reported by most investigators as the most common cause of the genesis of hypoxemia in PE. Correction of mismatching alveolar ventilation to cardiac output (CO) ratio (VA/Q) may occur from the redistribution of the perfusion towards ventilated areas or from a fall in ventilation of areas with limited or absent perfusion (3-6,8,10,13,14). Indeed, Levy and Simmons (15) observed that in PE there is a partial auto-regulation of the alveolar ventilation and perfusion at the lobar and segmental levels in order to maintain the equilibrium of the VA/Q (4,16). This phenomenon has only recently been confirmed with technology based on positron emission tomography scan that quantifies the redirection of the ventilation to alveoli with maintained perfusion, called effective or regional alveolar ventilation (VAeff) (14,17,18). Most studies attribute the redirection of alveolar ventilation to regional bronchoconstriction induced by alveolar hypocapnia (14,15,17,19,20). However, this redistribution of alveolar ventilation varying from nonexistent to highly significant has been frequently reported in the literature (8,9,15), probably because of the different approaches and models used (3).
A third element described in the pathogenesis of hypoxemia in PE is the reduction in PVO2 as a result of the fall in CO (8).
The aim of the present study was to determine the effective alveolar ventilation to perfusion ratio as a possible causal factor of hypoxemia in an experimental acute model of PE.
Material and Methods
The study was conducted in the Núcleo de Medicina e Cirurgia Experimental of the State University of Campinas (UNICAMP) and was approved by the Ethics Committee for Animal Research of the Institute of Biology, UNICAMP.
Seven Large-White pigs weighing 24.00 ± 0.6 kg were pretreated with 10 mg/kg intramuscular ketamine and subsequently anesthetized with 12 µg/kg fentanyl citrate and 25 mg/kg sodium thiopentate.
The animals were intubated and connected to a Servo 900B mechanical ventilator in a volume-controlled mode, with inspired oxygen fraction of 0.21 and minute volume and respiratory rate adjusted to maintain a carbon dioxide partial pressure in arterial blood (PaCO2) of about 40 mmHg. The tidal volume (VT) was approximately 350 mL and the positive pressure at the end of exhalation was adjusted to 5 cmH2O with decreasing inspiratory air flow.
A #5F-Swan-Ganz pulmonary artery catheter was introduced through the right femoral vein. A #6F-polyethylene catheter was introduced through the femoral artery and the tip was guided to the abdominal aorta. The correct location of the catheters was checked systematically by the analysis of the morphology of the pressure curves. Each catheter was filled with saline solution containing 5 units of heparin/mL and was connected to a Medex electromanometer (Hilliard, OH, USA). CO and blood pressure were recorded with a cardiac monitor (BESE, Belo Horizonte, MG, Brazil). All pressures were measured with the animals in the supine position and the zero reference point was set at the mid-thoracic line of the animal.
The blood pressure monitor was calibrated with a standard mercury column and CO was obtained by thermodilution, with the injection of 5% dextrose in water at a temperature of less than 5ºC.
The central temperature was obtained directly from the thermistor located at the tip of the pulmonary artery. Blood samples were collected simultaneously from the arterial and mixed venous lines and immediately analyzed with an IL-640 instrument (Instrumentation Laboratory, Lexington, MA, USA). Blood lactate was measured with an equipment that provides lactate values within 1 min from a single drop of arterial blood (Accusport, Boehringer Mannheim, Asta Medica, São Paulo, SP, Brazil).
The following hemodynamic parameters were measured and their variables calculated: CO, mean arterial pressure (MAP), pulmonary artery pressure (PAP), occluded pulmonary artery pressure, and heart rate. Capnography was carried out using the respiratory profile monitor DX-8100 (CO2 SMOPlus, Novametrix/Respironics, Cheshire, CT, USA). Analysis Plus software (Novametrix/Respironics) was used for the calculation of the physiological respiratory variables. The CO2SMOPlus pneumotachograph is based on the principle of differential manometry with a fixed resistance and is auto-calibrated.
One hour before embolization, 100 IU lyophilized bovine thrombin (Laboratory Instrumentation Company, Lexington, MA, USA) were added to 200 mL of blood removed from the jugular vein. After a period of 45 min the clotted blood was fragmented with a manual processor in order to obtain uniform fragments of clots measuring approximately 3 mm in diameter. These thrombi were filtered and suspended in saline solution and placed in a large syringe connected to a #14F-polyethylene catheter inserted into the left jugular vein of the animal.
The clots were injected in small volumes in order to reach the end point of a mean PAP (MPAP) two and a half times the MPAP recorded before embolism. The total volume of injected clots was 24.66 ± 4.27 mL and the average total time of clot injection was 20 min. Recordings of hemodynamic, capnographic, arterial and mixed venous blood gases, and blood lactate were carried out before embolization (baseline) and every 20 min starting at the end of clot injection. Four recordings were obtained, T0 (baseline) and T20, T40, and T60, at 20, 40, and 60 min, respectively, after MPAP established as the end point of embolization.
The Fowler method was used by the CO2SMOPlus to calculate the anatomical dead space (VDana). The physiological dead space (VD/VTphys) was calculated with the equation of Bohr-Enghoff (21):
VD/VTphys = (PaCO2 - PECO2)/PaCO2
where PECO2 is the partial pressure of mean CO2 in exhaled air.
The alveolar dead space (VDalv) was calculated as the difference between the physiological dead volume (VDphys, Bohr-Enghoff) (21) and the VDana.
VDalv = VDphys - VDana
Tidal alveolar volume (VTalv) was calculated as the simple difference between the VT and the VDphys.
VTalv = VT - VDphys
The VAeff was calculated as the simple difference between the VTalv and the VDphys.
VAeff = VTalv - VDphys
The partial pressure of oxygen in alveolar air (PAO2) was calculated from the equation of the alveolar air (22) with the formula:
PAO2 = PIO2 - (PaCO2/QR)
where PIO2 is the partial oxygen pressure on inspired air, PaCO2 was obtained from blood gas report and QR is the respiratory quotient considered to be fixed and equal to 0.8.
Systemic vascular resistance (SVR) was calculated by the equation SVR = MAP/CO and pulmonary vascular resistance (PVR) was calculated by the equation PVR = MPAP/CO.
Statistical analysis
Analysis of variance (ANOVA) for repeated measures with rank was used to compare the variables of interest for each group and for each time, with the level of significance set at 5% (P < 0.05).
Results
After embolization there was a significant fall in oxygen partial pressure in arterial blood (PaO2), PVO2 and PAO2, as shown in Figure 1.
Alveolar to arterial oxygen partial pressure gradient (P(A-a)O2) increased at T20 after embolization. However, it fell again at T60. Figure 2 shows a significant increase of PaCO2 and carbon dioxide partial pressure in mixed venous blood 20 min after embolism. Exhaled air at the end of the respiratory cycle (PetCO2) was significantly reduced at this time, returning to baseline at T40 (Figure 2). However, the arterial to alveolar carbon dioxide gradient (P(a-et)CO2) increased significantly and remained elevated up to T60.
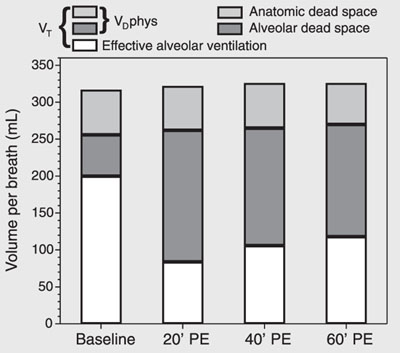
Figure 3 shows increases of VDalv and VDphys. VDana did not change. The VTalv remained unchanged throughout the experiment. On the other hand, at T20, VAeff exhibited a significant reduction despite a fixed minute volume, followed by a tendency to recovery.
The VAeff/Q ratio exhibited a significant reduction at T20 after embolization (Figure 4). However, at T40 and T60 these values tended to recover partially.
MPAP starting from a mean baseline value of 15.57 ± 2.23 mmHg reached a mean value of 46.71 ± 3.90 mmHg after embolization (T20), with no changes in CO. On the other hand, all animals presented severe tachycardia at T20. However, mean blood pressure did not change after embolization. Pulmonary vascular resistance increased significantly at T20, remaining elevated at T40 and T60 (Figure 5).
Partial pressure of oxygen in alveolar air (PAO2), in arterial blood (PaO2) and in venous blood (PvO2) during experimental pulmonary embolism (PE) in pigs. Data are reported as means ± SD for 7 pigs.
Carbon dioxide partial pressure in arterial blood (PaCO2) and in venous blood (PvCO2), and end tidal exhaled air (PetCO2) during experimental pulmonary embolism (PE) in pigs. Data are reported as means ± SD for 7 pigs.
Respiratory variation in volume per breath during experimental pulmonary embolism (PE) in pigs. Data are reported as means for 7 pigs. VT = tidal volume; VDphys = physiological dead space.
Effective alveolar ventilation per breath (VAeff) and effective alveolar ventilation to cardiac output ratio (VAeff/Q) during experimental pulmonary embolism (PE) in pigs. Data are reported as means ± SD for 7 pigs.
Pulmonary vascular resistance (PVR) and mean pulmonary artery pressure (MPAP) during experimental pulmonary embolism (PE) in pigs. Data are reported as means ± SD for 7 pigs.
Discussion
Blockade of pulmonary perfusion by clots results in ventilation of non-perfused alveoli, characterizing wasted ventilation, conventionally named VDalv. The increase in VDalv subtracts a considerable fraction of alveolar ventilation, leading to reduction of VAeff (15).
The present data show that VDana was unchanged, whereas VDphys and VDalv increased in an expressive way as a result of blood clots impairing the pulmonary circulation of the animals (2,13).
Since no increase in VDana was detected, the expressive increase in VDphys could be exclusively attributed to the increase in VDalv. The significant increase in PaCO2 after embolization was attributed to the abolished reflex hyperventilation normally triggered by hypercapnia, because the animals were sedated and mechanically ventilated (2,15).
It could be argued that the reduction of VDalv, the P(a-et)CO2 gradient and the improvement of VAeff and arterial oxygenation at T40 and T60 were caused by spontaneous thrombolysis. If this was the case, a reduction in pulmonary vascular resistance should be expected. Actually, pulmonary vascular resistance increased at T20 and remained elevated at T40 and T60 (Figure 5). This fact can only be attributed to vasoconstriction induced by the low PAO2 values (Figure 1) since it has been shown that PAO2 values below 70 mmHg trigger marked vasoconstriction, up to the point where blood flow is abolished (8,15,23,24).
On the other hand, the reduction of PetCO2 was reversed at T40. This behavior was interpreted as redistribution of alveolar ventilation and as relative reperfusion of the vascular bed due to the reduction of VDalv at T40 (Figure 2) despite elevated values compared to baseline.
In a study of the kinetics of carbon dioxide pulmonary artery occlusion in dogs with subsequent reperfusion, Anderson and Breen (13) stated that the return of PetCO2 to baseline values is indicative of pulmonary perfusion and, indeed, in the present study PetCO2 returned to baseline levels at T40 (Figure 2). However, this finding cannot be attributed exclusively to thrombolysis since PaO2 and PaCO2 did not normalize.
As can be seen in Figure 1, the animals presented an important fall in PAO2 and PaO2 which was much more pronounced than expected, causing only an increase in alveolar carbon dioxide. For this reason, a second mechanism must be responsible for the important hypoxemia observed, such as pulmonary shunt or mismatch between ventilation and perfusion (23).
Venous admixture (Qva/Qt) increased significantly. However, this variable does not discriminate venous mixture from true shunt. Despite suggestions that alterations in surfactant synthesis due to pulmonary embolism can induce alveolar collapse (8,25), it is hard to believe that these alterations would occur as early as 20 min after PE. Also, the true shunt could hardly be attributed to alveolar exclusion as observed in acute respiratory distress syndrome or in acute pulmonary edema. For these reasons, it is more sensible to attribute the increase of the Qva/Qt ratio to a VA/Q imbalance.
In this model of PE, redistribution of perfusion must have necessarily occurred since there were non-perfused areas in the pulmonary circulation and CO was unchanged.
In this experimental model, the ventilated, but non-perfused alveoli increased VDalv from 56.38 ± 22.47 at baseline to 178.09 ± 37.46 mL at T20. Actually, the increase in dead space per se does not induce hypoxemia. For this reason, hypoxemia is attributed to a significant alteration of the VA/Q ratio (3,5,10,14,15,17,25), with alveolar ventilation being slightly increased but with perfusion being markedly increased.
The effective alveolar ventilation is defined as the ventilation in contact with the pulmonary circulation (26). Although the term "effective alveolar ventilation" is not currently used in respiratory physiology textbooks, Levy and Simmons (15), in 1974, introduced this term to characterize exclusively the ventilation of areas with maintained or increased perfusion. The increase in VAeff with time (Figures 3 and 4) was attributed to hypocapnic bronchoconstriction because the authors showed that a group of animals that inhaled 3% CO2 after pulmonary embolism did not exhibit the expected increase in VAeff (15).
In the present study, the VTalv remained unchanged because the animals were on mechanical ventilation with a fixed minute volume. The non-perfused pulmonary areas were not anatomically quantified after embolization. However, in the sequential evaluation, a significant increase in VAeff at T40 and T60 was observed when compared to the VAeff at T20 after embolism. This evidence suggests that the mechanism of hypocapnic bronchoconstriction did occur and that the alveolar ventilation was effectively redirected to units of effective gas exchange. This recovery of VAeff was associated with a significant improvement in the VAeff/Q ratio as well as arterial oxygenation at T40 and T60 (Figures 1 and 4). The fall in the P(A-a)O2 at T40 and T60 corroborates the increase in VAeff and VAeff/Q induced by redirection of ventilation. Similarly, the normalization of PetCO2 as early as 40 min after embolization expresses the redirectioning of alveolar ventilation by the hypocapnic alveolar bronchoconstriction (Figure 2). However, the increase in P(a-et)CO2 (Figure 2) is essentially attributed to the persistent elevation of PaCO2 induced by a fixed minute volume.
Vidal Melo et al. (14), after injecting autologous clots in six mechanically ventilated sheep, confirmed by positron emission tomography scan that there was an increase in regional alveolar ventilation in pulmonary areas that remained perfused after embolization, as an expression of the redistribution of VT mediated by the hypocapnic bronchoconstriction. The quantity and quality of the clots used in the experimental studies may influence the duration and intensity of bronchoconstriction. Fresh clots may liberate serotonin because of the presence of thrombin. Robinson et al. (27) showed that serotonin has an important bronchoconstrictor effect when pulmonary embolization is induced with fresh clots. Levy and Simmons (15) observed a bronchoconstriction of 30-min duration after injection of 30 mL of fresh clots. In our study, bronchoconstriction occurred up to 60 min after embolization, as suggested by the progressive increase in effective alveolar ventilation. This finding is attributed to the large quantity of clots (24.66 ± 4.27 mL) and to the added lyophilized bovine thrombin.
In the present study, VDalv as well as P(a-et)CO2 increased significantly at T20 (56.38 ± 22.47 to 178.09 ± 37.46 mL, and 1.94 ± 1.36 to 37.61 ± 12.79 mmHg, respectively), but decreased at T40 and T60. This was also observed by Skimming et al. (28) in 11 mechanically ventilated sheep with induced acute pulmonary lesion, with the authors concluding that P(a-et)CO2 was a good predictor of VDalv. Similarly, Moreira et al. (29), comparing two experimental situations, i.e., PE and hemorrhagic shock, demonstrated a significant increase of P(a-et)CO2 only in PE, which was associated with increased VDalv as measured by volumetric capnography. The increase in P(a-et)CO2 was significant and sufficiently specific to suggest the diagnosis of PE.
Based on our data obtained in animals under mechanical ventilation and submitted to pulmonary embolism by injection of autologous clots, we conclude that the severe arterial hypoxemia observed must be attributed to the reduction of both VAeff and the VAeff/Q ratio. These variables, as well as the arterial hypoxemia, tended to recover with time, but did not return to baseline levels even after 60 min of embolism. This behavior is attributed to the redistribution of the alveolar ventilation induced by hypocapnic bronchoconstriction.
References
1. Wood KE. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest 2002; 121: 877-905.
2. Goldhaber SZ, Elliott CG. Acute pulmonary embolism: Part I: epidemiology, pathophysiology, and diagnosis. Circulation 2003; 108: 2726-2729.
3. Altemeier WA, Robertson HT, McKinney S, Glenny RW. Pulmonary embolization causes hypoxemia by redistributing regional blood flow without changing ventilation. J Appl Physiol 1998; 85: 2337-2343.
4. Brimioulle S, Lejeune P, Naeije R. Effects of hypoxic pulmonary vasoconstriction on pulmonary gas exchange. J Appl Physiol 1996; 81: 1535-1543.
5. Huet Y, Lemaire F, Brun-Buisson C, Knaus WA, Teisseire B, Payen D, et al. Hypoxemia in acute pulmonary embolism. Chest 1985; 88: 829-836.
6. Manier G, Castaing Y, Guenard H. Determinants of hypoxemia during the acute phase of pulmonary embolism in humans. Am Rev Respir Dis 1985; 132: 332-338.
7. Kasinski N, Afonso JE, Ferreira C, dos Santos ML, Ratto OR, Barcellini A. Pulmonary embolism with autologous blood clot. Experimental study in dogs. Arq Bras Cardiol 1980; 34: 203-210.
8. Dantzker DR, Bower JS. Alterations in gas exchange following pulmonary thromboembolism. Chest 1982; 81: 495-501.
9. Santolicandro A, Prediletto R, Fornai E, Formichi B, Begliomini E, Giannella-Neto A, et al. Mechanisms of hypoxemia and hypocapnia in pulmonary embolism. Am J Respir Crit Care Med 1995; 152: 336-347.
10. Póvoa R, Kasinski N. Hipertensão pulmonar no tromboembolismo pulmonar. Rev Soc Cardiol Estado São Paulo 2000; 5: 590-598.
11. Jatene FB, Bernardo WM. Isquemia pulmonar embólica - aspectos clínicos e experimentais. Rev Assoc Med Bras 2003; 49: 342-348.
12. Wilson JE III, Pierce AK, Johnson RL Jr, Winga ER, Harrell WR, Curry GC, et al. Hypoxemia in pulmonary embolism, a clinical study. J Clin Invest 1971; 50: 481-491.
13. Anderson CT, Breen PH. Carbon dioxide kinetics and capnography during critical care. Crit Care 2000; 4: 207-215.
14. Vidal Melo MF, Harris RS, Layfield D, Musch G, Venegas JG. Changes in regional ventilation after autologous blood clot pulmonary embolism. Anesthesiology 2002; 97: 671-681.
15. Levy SE, Simmons DH. Redistribution of alveolar ventilation following pulmonary thromboembolism in the dog. J Appl Physiol 1974; 36: 60-68.
16. Naeije R, Brimioulle S. Physiology in medicine: importance of hypoxic pulmonary vasoconstriction in maintaining arterial oxygenation during acute respiratory failure. Crit Care 2001; 5: 67-71.
17. Severinghaus JW, Swenson EW, Finley TN, Lategola MT, Williams J. Unilateral hypoventilation produced in dogs by occluding one pulmonary artery. J Appl Physiol 1961; 16: 53-60.
18. Vidal Melo MF, Layfield D, Harris RS, O'Neill K, Musch G, Richter T, et al. Quantification of regional ventilation-perfusion ratios with PET. J Nucl Med 2003; 44: 1982-1991.
19. Simon BA, Tsuzaki K, Venegas JG. Changes in regional lung mechanics and ventilation distribution after unilateral pulmonary artery occlusion. J Appl Physiol 1997; 82: 882-891.
20. Simon BA, Zanaboni PB, Nyhan DP. Effect of hypoxia on respiratory system impedance in dogs. J Appl Physiol 1997; 83: 451-458.
21. Enghoff H. Volumem Inefecax. Bemerkungen zur Frage dês schaedlichen Raumes. Uppsala Läk For Förh 1938; 44: 191-218.
22. Terzi RGG, Dragosavac D. Ventilação mecânica básica. In: Carvalho CRR (Editor), Monitorização do intercâmbio gasoso pulmonar no paciente submetido à ventilação mecânica. São Paulo: Atheneu; 2000. p 189-213.
23. West JB. Fisiologia respiratória. In: West J (Editor), Ventilação - como o gás chega aos alvéolos. São Paulo: Editora Manole; 2002. p 11-20.
24. Marshall BE, Hanson CW, Frasch F, Marshall C. Role of hypoxic pulmonary vasoconstriction in pulmonary gas exchange and blood flow distribution. 2. Pathophysiology. Intensive Care Med 1994; 20: 379-389.
25. Baruzzi ACA, Knobel E, Kasinski N, Junior CJF. Condutas no paciente grave. In: Knobel E (Editor), Tromboembolismo pulmonar. São Paulo: Atheneu; 1994. p 8-19.
26. Stenqvist O, Olausson M, Karlsen KL. Luxury lung perfusion in end-stage liver disease during liver transplantation. Acta Anaesthesiol Scand 1999; 43: 447-451.
27. Robinson AE, Puckett CL, Green JD, Silver D. In vivo demonstration of small-airway bronchoconstriction following pulmonary embolism. Radiology 1973; 109: 283-286.
28. Skimming JW, Banner MJ, Spalding HK, Jaeger MJ, Burchfield DJ, Davenport PW. Nitric oxide inhalation increases alveolar gas exchange by decreasing deadspace volume. Crit Care Med 2001; 29: 1195-1200.
29. Moreira MM, Ferreira ELA, Terzi RGG, Silva WA, Moraes AC, Metze K. Avaliação do espaço morto no tromboembolismo pulmonar e no choque hemorrágico experimentais. Rev Bras Terap Intensiva 2004; 17: 6-14.
Address for correspondence: J.H.T. Ferreira, Rua João Bissoto Filho, 99, Bloco 15, Apto. 21, 13275-410 Valinhos, SP, Brasil. Fax: +55-19-3233-2969. E-mail: juterra2000@yahoo.com.br or rggterzi@hotmail.com
Publication supported by FAPESP. Received November 24, 2005. Accepted June 12, 2006.
- 1. Wood KE. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest 2002; 121: 877-905.
- 2. Goldhaber SZ, Elliott CG. Acute pulmonary embolism: Part I: epidemiology, pathophysiology, and diagnosis. Circulation 2003; 108: 2726-2729.
- 3. Altemeier WA, Robertson HT, McKinney S, Glenny RW. Pulmonary embolization causes hypoxemia by redistributing regional blood flow without changing ventilation. J Appl Physiol 1998; 85: 2337-2343.
- 4. Brimioulle S, Lejeune P, Naeije R. Effects of hypoxic pulmonary vasoconstriction on pulmonary gas exchange. J Appl Physiol 1996; 81: 1535-1543.
- 5. Huet Y, Lemaire F, Brun-Buisson C, Knaus WA, Teisseire B, Payen D, et al. Hypoxemia in acute pulmonary embolism. Chest 1985; 88: 829-836.
- 6. Manier G, Castaing Y, Guenard H. Determinants of hypoxemia during the acute phase of pulmonary embolism in humans. Am Rev Respir Dis 1985; 132: 332-338.
- 7. Kasinski N, Afonso JE, Ferreira C, dos Santos ML, Ratto OR, Barcellini A. Pulmonary embolism with autologous blood clot. Experimental study in dogs. Arq Bras Cardiol 1980; 34: 203-210.
- 8. Dantzker DR, Bower JS. Alterations in gas exchange following pulmonary thromboembolism. Chest 1982; 81: 495-501.
- 9. Santolicandro A, Prediletto R, Fornai E, Formichi B, Begliomini E, Giannella-Neto A, et al. Mechanisms of hypoxemia and hypocapnia in pulmonary embolism. Am J Respir Crit Care Med 1995; 152: 336-347.
- 10. Póvoa R, Kasinski N. Hipertensão pulmonar no tromboembolismo pulmonar. Rev Soc Cardiol Estado São Paulo 2000; 5: 590-598.
- 11. Jatene FB, Bernardo WM. Isquemia pulmonar embólica - aspectos clínicos e experimentais. Rev Assoc Med Bras 2003; 49: 342-348.
- 12. Wilson JE III, Pierce AK, Johnson RL Jr, Winga ER, Harrell WR, Curry GC, et al. Hypoxemia in pulmonary embolism, a clinical study. J Clin Invest 1971; 50: 481-491.
- 13. Anderson CT, Breen PH. Carbon dioxide kinetics and capnography during critical care. Crit Care 2000; 4: 207-215.
- 14. Vidal Melo MF, Harris RS, Layfield D, Musch G, Venegas JG. Changes in regional ventilation after autologous blood clot pulmonary embolism. Anesthesiology 2002; 97: 671-681.
- 15. Levy SE, Simmons DH. Redistribution of alveolar ventilation following pulmonary thromboembolism in the dog. J Appl Physiol 1974; 36: 60-68.
- 16. Naeije R, Brimioulle S. Physiology in medicine: importance of hypoxic pulmonary vasoconstriction in maintaining arterial oxygenation during acute respiratory failure. Crit Care 2001; 5: 67-71.
- 17. Severinghaus JW, Swenson EW, Finley TN, Lategola MT, Williams J. Unilateral hypoventilation produced in dogs by occluding one pulmonary artery. J Appl Physiol 1961; 16: 53-60.
- 18. Vidal Melo MF, Layfield D, Harris RS, O'Neill K, Musch G, Richter T, et al. Quantification of regional ventilation-perfusion ratios with PET. J Nucl Med 2003; 44: 1982-1991.
- 19. Simon BA, Tsuzaki K, Venegas JG. Changes in regional lung mechanics and ventilation distribution after unilateral pulmonary artery occlusion. J Appl Physiol 1997; 82: 882-891.
- 20. Simon BA, Zanaboni PB, Nyhan DP. Effect of hypoxia on respiratory system impedance in dogs. J Appl Physiol 1997; 83: 451-458.
- 21. Enghoff H. Volumem Inefecax. Bemerkungen zur Frage dês schaedlichen Raumes. Uppsala Läk For Förh 1938; 44: 191-218.
- 22. Terzi RGG, Dragosavac D. Ventilação mecânica básica. In: Carvalho CRR (Editor), Monitorização do intercâmbio gasoso pulmonar no paciente submetido à ventilação mecânica São Paulo: Atheneu; 2000. p 189-213.
- 23. West JB. Fisiologia respiratória. In: West J (Editor), Ventilação - como o gás chega aos alvéolos São Paulo: Editora Manole; 2002. p 11-20.
- 24. Marshall BE, Hanson CW, Frasch F, Marshall C. Role of hypoxic pulmonary vasoconstriction in pulmonary gas exchange and blood flow distribution. 2. Pathophysiology. Intensive Care Med 1994; 20: 379-389.
- 25. Baruzzi ACA, Knobel E, Kasinski N, Junior CJF. Condutas no paciente grave. In: Knobel E (Editor), Tromboembolismo pulmonar São Paulo: Atheneu; 1994. p 8-19.
- 26. Stenqvist O, Olausson M, Karlsen KL. Luxury lung perfusion in end-stage liver disease during liver transplantation. Acta Anaesthesiol Scand 1999; 43: 447-451.
- 27. Robinson AE, Puckett CL, Green JD, Silver D. In vivo demonstration of small-airway bronchoconstriction following pulmonary embolism. Radiology 1973; 109: 283-286.
- 28. Skimming JW, Banner MJ, Spalding HK, Jaeger MJ, Burchfield DJ, Davenport PW. Nitric oxide inhalation increases alveolar gas exchange by decreasing deadspace volume. Crit Care Med 2001; 29: 1195-1200.
- 29. Moreira MM, Ferreira ELA, Terzi RGG, Silva WA, Moraes AC, Metze K. Avaliação do espaço morto no tromboembolismo pulmonar e no choque hemorrágico experimentais. Rev Bras Terap Intensiva 2004; 17: 6-14.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
27 Feb 2008 -
Date of issue
Sept 2006
History
-
Accepted
12 June 2006 -
Received
24 Nov 2005