Abstract
Occupational therapy (OT) is a profession concerned with promoting health and well-being through occupation, by enabling handicapped people to participate in the activities of everyday life. OT is part of the clinical rehabilitation of progressive genetic neurodegenerative diseases such as spinocerebellar ataxias; however, its effects have never been determined in these diseases. Our aim was to investigate the effect of OT on both physical disabilities and depressive symptoms of spinocerebellar ataxia type 3 (SCA3) patients. Genomically diagnosed SCA3 patients older than 18 years were invited to participate in the study. Disability, as evaluated by functional independence measurement and Barthel incapacitation score, Hamilton Rating Scale for Depression, and World Health Organization Quality of Life questionnaire (WHOQOL-BREF), was determined at baseline and after 3 and 6 months of treatment. Twenty-six patients agreed to participate in the study. All were treated because OT prevents blinding of a control group. Fifteen sessions of rehabilitative OT were applied over a period of 6 months. Difficult access to food, clothing, personal hygiene, and leisure were some of the main disabilities focused by these patients. After this treatment, disability scores and quality of life were stable, and the Hamilton scores for depression improved. Since no medication was started up to 6 months before or during OT, this improvement was related to our intervention. No association was found between these endpoints and a CAG tract of the MJD1 gene (CAGn), age, age of onset, or neurological scores at baseline (Spearman test). Although the possibly temporary stabilization of the downhill disabilities as an effect of OT remains to be established, its clear effect on depressive symptoms confirms the recommendation of OT to any patient with SCA3 or spinocerebellar ataxia.
Spinocerebellar ataxia 3; Occupational therapy; Rehabilitation; Depression; Machado-Joseph disease; Polyglutamine diseases
Braz J Med Biol Res, June 2010, Volume 43(6) 537-542
Occupational therapy in spinocerebellar ataxia type 3: an open-label trial
R.C.R. Silva1,4, J.A.M. Saute5, A.C.F. Silva5, A.C.O. Coutinho4, M.L. Saraiva-Pereira1,2,5 and  Correspondence and Footnotes
Correspondence and Footnotes
 L.B. Jardim1,3,5
L.B. Jardim1,3,5
1Programa de Pós-Graduação em Ciências Médicas, 2Departamento de Bioquímica, 3Departamento de Medicina Interna, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brasil
4Serviço de Reabilitação, 5Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Porto Alegre, RS, Brasil
 Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Occupational therapy (OT) is a profession concerned with promoting health and well-being through occupation, by enabling handicapped people to participate in the activities of everyday life. OT is part of the clinical rehabilitation of progressive genetic neurodegenerative diseases such as spinocerebellar ataxias; however, its effects have never been determined in these diseases. Our aim was to investigate the effect of OT on both physical disabilities and depressive symptoms of spinocerebellar ataxia type 3 (SCA3) patients. Genomically diagnosed SCA3 patients older than 18 years were invited to participate in the study. Disability, as evaluated by functional independence measurement and Barthel incapacitation score, Hamilton Rating Scale for Depression, and World Health Organization Quality of Life questionnaire (WHOQOL-BREF), was determined at baseline and after 3 and 6 months of treatment. Twenty-six patients agreed to participate in the study. All were treated because OT prevents blinding of a control group. Fifteen sessions of rehabilitative OT were applied over a period of 6 months. Difficult access to food, clothing, personal hygiene, and leisure were some of the main disabilities focused by these patients. After this treatment, disability scores and quality of life were stable, and the Hamilton scores for depression improved. Since no medication was started up to 6 months before or during OT, this improvement was related to our intervention. No association was found between these endpoints and a CAG tract of the MJD1 gene (CAGn), age, age of onset, or neurological scores at baseline (Spearman test). Although the possibly temporary stabilization of the downhill disabilities as an effect of OT remains to be established, its clear effect on depressive symptoms confirms the recommendation of OT to any patient with SCA3 or spinocerebellar ataxia.
Key words: Spinocerebellar ataxia 3; Occupational therapy; Rehabilitation; Depression; Machado-Joseph disease; Polyglutamine diseases
Introduction
Spinocerebellar ataxia 3 (SCA3), also known as Machado-Joseph disease, is a rare neurodegenerative disease caused by expansions of a CAG tract of the MJD1 gene (1-3). The expanded allele is dominant, and there is an important correlation of the repeat amplification with both symptom severity and age at onset in affected individuals. There is no treatment.
SCA3 affects at least 3:100,000 individuals in the Brazilian population (4). SCA3 is a highly disabling disease, which imposes a severe burden on the patients and their families. Clinical manifestations usually start during adulthood, with a mean (± SD) age at onset of 32 ± 12 years (5). Patients end up confined to a wheelchair and later become bedridden (6). Age at onset distribution is very wide and ranges between 5 and 73 years (6). Median survival time after onset is 21 years (7).
The disease is related to neuronal loss and neuronal intranuclear inclusions, detected mainly in the dentate nucleus of the cerebellum, the nucleus dorsalis of Clarke in the spinal cord, cranial motor nerve nuclei, pontine nuclei, substantia nigra, and the lenticular fasciculus of the globus pallidus (8-11). Clinical manifestations usually start with cerebellar ataxia affecting gait, limb movements, speech articulation, and deglutition. Patients also present a variety of other dysfunctions, including pyramidal involvement; a supranuclear, progressive external ophthalmoplegia with limitation of upward gaze and convergence; extrapyramidal signs, including dystonia, rigidity, bradykinesia, and even a full parkinsonian syndrome; lower motor neuron disease, with fasciculation and amyotrophy; sensitive loss; eyelid retraction, contraction fasciculation, weight loss, and sleep disorder (6). All of these findings lead to a progressive burden and incapacitation. In addition, depressive symptoms are rather frequent and may be related to the inexistence of an effective treatment (12). In SCA3, depressive scores have been associated with the level of incapacitation, conditions that probably reinforce each other.
To our knowledge, very few studies of rehabilitation interventions in SCAs in general and in SCA3 in particular have appeared in the literature. Among rehabilitation techniques, occupational therapy (OT) aims to adapt a particular patient to the activities of his/her daily living in order to obtain the maximum possible independence. OT is a common clinical practice, although its effects have never been measured in patients with SCA. In order to identify the role of OT in SCA3, the present study describes the disabilities associated with the disease, the effect of occupational therapy on these disabilities, on depressive symptoms and on quality of life, and the possible associations of these endpoints with risk factors, such as a CAG tract of the MJD1 gene (CAGn) and age of onset.
Material and Methods
Genomically diagnosed SCA3 patients older than 18 years were invited to participate. The inclusion criterion was independent gait on neurological examination. Patients who had started any therapy less than 6 months before or who had previously been on OT were excluded. The present study was approved by the Grupo de Pesquisa e de Pós-Graduação do Hospital de Clínicas de Porto Alegre (GPPG, process #05-254), and all patients gave written informed consent before participating.
After the patients agreed to participate in the study, a structured interview, which included four instruments to measure endpoints, was performed. The disability scores included the functional independence measurement (FIM) in its Portuguese version (13,14), and Barthel Incapacitation scores (15). FIM scores were classified as follows: under 18, total dependence; 18 to 60, 50% dependence; 61 to 103, 25% dependence, and over 104, preserved independence. Barthel scores were classified as follows: 0 to 45, severe motor disability; 46 to 75, moderate disability; 76 to 99, mild disability, and 100, no disability. Depressive symptoms were measured by the Hamilton Rating Scale for Depression (16). Scores over 25, between 18 and 24, and between 17 and 7 were associated with severe, moderate and mild depression, respectively. Quality of life was investigated with the Portuguese version of the World Health Organization Quality of Life questionnaire (WHOQOL-BREF) (17,18). At baseline, the Neurological Examination Score for Spinocerebellar Ataxia (NESSCA) (19) and the Scale for Assessment and Rating of Ataxia (SARA) score (20) were performed by previously trained observers (JAMS and ACFS).
The OT frame of reference followed the rehabilitative (compensatory) model (21). OT intervention consisted of weekly sections of 40 min during the first 3 months, followed by monthly sections during another 3 months. In the first interview, the most significant functional limitations in the patient’s life were identified according to patient’s opinion. The following items were considered: access to food, clothing, bathroom use, and personal hygiene; important activities for the individual’s economic support; leisure activities, and the ways the patient interacts and meets with his/her social and affective circles. Both patient and occupational therapist elected priorities, including viable objectives only. As a result, the patient and the therapist agreed upon a practical plan; if necessary, modifications were made during follow-up. In short, interventions followed the clinical practice of OT. The endpoint instruments were applied again at 3 and 6 months after the beginning of OT treatment. Patients were maintained thereafter on bi-monthly follow-ups.
Hamilton, Barthel, FIM, and WHOQOL scores (the endpoints) at baseline and after either 3 or 6 months on OT were compared using the paired Student t-test if variables showed normal distribution; baseline scores were compared with the final scores of the same individual. Non-parametric variables were tested by the Wilcoxon U-test or the Kruskal-Wallis test. Possible associations between endpoints were tested using the Spearman correlation test. Correlations were also tested between endpoints and the following risk factors by the Spearman test: age, age of onset, CAGn, NESSCA, and SARA scores. Bonferroni corrections were made due to the use of multiple tests. The level of significance was set at 0.05.
Results
Twenty-six individuals entered the study. They all had the same geographical and urban origin and lived in Rio Grande do Sul, Brazil, where SCA3 cases seem to be limited to persons of Azorean ancestry and moreover to the ancestral haplotype A-C-A (22). Their clinical and genomic characteristics are summarized in Table 1. The group of patients showed on average mild depressive symptoms. The disability scores of these patients were also mild, although neurological findings were substantial (NESSCA and SARA). Quality of life was on average moderately compromised, and there was no skewness of the results.
Three individuals were lost to follow-up: 1 patient did not return to the third monthly visit, and the other 2 patients did not return at the end of the study. Age, schooling, and baseline NESSCA, SARA, FIM, Barthel, and Hamilton scores were similar for both the lost and remaining patients. Although non-significant, losses showed a trend towards having a longer CAGn when compared to the general sample (78 vs 74) and consequently earlier ages at onset (28.6 ± 13.3 vs 37.1 ± 10 years). The quality of life of cases lost to follow-up also tended to be worse than that of the general group (45.8 ± 14.4 vs 58.1 ± 9.6; P = 0.06).
Follow-up of depressive symptoms
Hamilton scores (mean ± SD) for depression obtained for the 23 patients who completed the study were 8.65 ± 6.6 and 6.04 ± 6.2 before and after 6 months of OT treatment, respectively. This improvement was not only significant (P < 0.0001, paired t-test), but the mean (after trial) also reached normal values. Individual scores are presented in Figure 1.
The difference between the 6th month score and baseline score, hereafter referred to as ∆ Hamilton, was not related to the independent variables under study (age, age at onset, schooling, CAGn, SARA, and NESSCA scores) or to the other endpoints measured (Barthel, WHOQOL, and FIM) at baseline or at the patients own progression slopes.
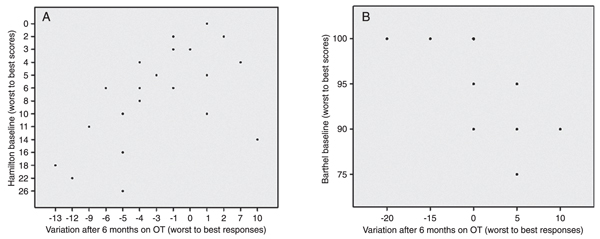
Individuals with higher depressive scores at baseline showed apparently better responses to treatment (OT) after 6 months (Figure 2A; r = -0.619, P < 0.002, Spearman).
Follow-up of FIM and Barthel scores for disability
The incapacities most referred to by patients were difficulty in dynamic balance resulting in walking deficits, difficulty in word articulation, and difficulty in handling tableware and pens.
Mean ± SD scores for disability did not change after 6 months of OT: FIM scores were 120.17 ± 4.8 and 120.26 ± 6.5, and Barthel scores were 96.9 ± 5.9 and 96.9 ± 6.3 at baseline and after 6 months. When the group was stratified according to CAGn (two groups of patients, with cut-off at 73 CAGs) and to disease duration (cut-off at 5 years of disease duration), no differences were found in FIM progression.
Some individuals actually got worse after 6 months of follow-up. Regarding Barthel scores, an inverse relation was observed between response (∆ Barthel) and baseline; the better the baseline, the worse the response after 6 months. This phenomenon is illustrated in Figure 2B.
Follow-up of quality of life
Mean ± SD scores of global WHOQOL did not change after 6 months of OT; they were 58.1 ± 9.6 and 58.1 ± 14.4 at baseline and after 6 months (paired t-test). Since patients lost to follow-up tended to have worse WHOQOL and in order to test if their exclusion could have biased these results, we also compared the WHOQOL of the overall sample at baseline (26 cases) to that of the remaining sample at 6 months (23 cases). No differences were found.
Actually, 4 patients improved according to WHOQOL, 15 remained the same, and 4 worsened at the end of observation. When these three subgroups were analyzed, no differences were found in their independent variables or in the other endpoints studied.
Progression of Hamilton Depression Scores for each individual under trial. Scores were obtained at baseline and after 3 and 6 months on occupational therapy.
Relation between baseline scores and response after 6 months on occupational therapy of (A) Hamilton Depression Rating Scale and (B) Barthel Incapacitation scores. In both graphs, the x-axis represents the difference between the 6 months and the baseline scores.
Discussion
OT is currently part of common clinical practice, although its effects have rarely been measured. Since interventions are individually tailored in any rehabilitation therapy but especially in OT, the variability of interventions or their qualitative nature can partially explain the lack of studies on their effects. OT is particularly important in progressively incapacitating diseases, especially those without any known treatment. The objective of OT is to improve abilities and capacities of daily living in handicapped individuals. And due to the progressive nature of neurodegenerative diseases, OT should not only be a permanent management but also change as disease progresses.
Studies on the impact of OT on SCAs in general are lacking. Two open-label trials on SCA2 studied the impact of physical training and of group psychotherapy (23,24). Both studies reported favorable follow-ups, but intervention, disease under study and endpoints were diverse, and therefore their results are hardly comparable to ours. Expert opinions are also rare: in a review article, neuroanatomical characteristics were analyzed regarding rehabilitative choices in SCA2 and SCA3 (25).
Since we routinely indicate OT to our patients, we tried to improve our knowledge about the role of this rehabilitation technique. Our challenge started when we tried to figure out what the specific effect of OT would be, for instance, on capacities and abilities, social adaptation, or personal adaptation to the disease. The existing instruments used to measure these endpoints are multiple, and there is no unique severity score with all these domains. Because of this, we decided to test disabilities, quality of life, and depressive manifestations. Our second challenge was the study design, and we decided for an open trial. This design was chosen for two main reasons. First, OT is an acclaimed management, which is no longer prone to randomized studies. Second, blinding OT would be rather impossible.
Our results indicate that OT improved the Hamilton scores for depression of SCA3 patients. This response to OT was not related to the independent variables under study, i.e., gender, age, age at onset, schooling, CAGn, NESSCA, and SARA scores. Moreover, improvement of depression did not correlate with the other endpoints.
Incapacitation scores - Barthel score and FIM - and quality of life were stable throughout this study. However, since the natural history of these parameters in SCA3 is unknown, interpretation of these data was not possible. This is a drawback of this study design, where no control group was monitored. When the natural history of a progressive disorder like SCA3 is unknown, open trials do not allow stabilizations to be interpreted as positive results. Stable states may theoretically be related to the natural course of a disease. Only improvements can be seen as definite results of open trials, in relentless diseases like SCA3.
Although the paired t-test did not detect any statistically significant differences in Barthel or FIM after 6 months of observation, a careful exam of the data revealed a sad reality. Baseline Barthel had an inverse correlation with responses, or ∆. In other words, patients with the best functional independence parameters at the beginning of the study tended to get worse after 6 months. This was clearly the result of the natural course of the disease.
The observation started with 26 cases; 3 dropped out, but there was apparently no bias due to these losses. In any case, it is possible that patients with more social networks and consequently better quality of life were inadvertently selected. Keeping in mind that this sample was limited to independent ambulatory patients, we still believe that this group was representative of SCA3 patients with an A-C-A ancestry, since general parameters such as age, age at onset, gender, CAGn ranges, and NESSCA scores were similar to those found in previous studies on these populations (5,19).
OT improved the depressive symptoms of SCA3 patients, which are an important clinical problem already shown to be common among these patients (12). Organic or reactive depressive symptoms can affect 33.5% of SCA3 individuals. The relation between depressive scores and incapacitation levels observed in previous studies was not found in the present sample (12). We speculate that our lack of association was due to the recruitment of patients with relatively shorter disease duration and lower impact on their capabilities. Of course, incapacitation and depression may reinforce each other. Depressive symptoms should be looked for in SCA3 and probably in other SCAs in order to offer appropriate support to these individuals. We have already reported positive results of fluoxetine treatment on depression in these patients (26), and now a similar effect concerning OT was found. Comparisons of costs and effects of both treatments (OT and pharmacological) are prevented, due to the lack of data about them.
Although the possibly temporary stabilization of the downhill disabilities as an effect of OT remains to be established, the clear effect on depressive symptoms is enough to confirm the recommendation of OT to any given patient with SCA3 or a spinocerebellar ataxia.
References
1. Takiyama Y, Nishizawa M, Tanaka H, Kawashima S, Sakamoto H, Karube Y, et al. The gene for Machado-Joseph disease maps to human chromosome 14q. Nat Genet 1993; 4: 300-304.
2. Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 1994; 8: 221-228.
3. Bauer PO, Kotliarova SE, Matoska V, Musova Z, Hedvicakova P, Boday A, et al. Fluorescent multiplex PCR - fast method for autosomal dominant spinocerebellar ataxias screening. Genetika 2005; 41: 830-837.
4. Prestes PR, Saraiva-Pereira ML, Silveira I, Sequeiros J, Jardim LB. Machado-Joseph disease enhances genetic fitness: a comparison between affected and unaffected women and between MJD and the general population. Ann Hum Genet 2008; 72: 57-64.
5. Jardim LB, Pereira ML, Silveira I, Ferro A, Sequeiros J, Giugliani R. Machado-Joseph disease in South Brazil: clinical and molecular characterization of kindreds. Acta Neurol Scand 2001; 104: 224-231.
6. Sequeiros J, Coutinho P. Epidemiology and clinical aspects of Machado-Joseph disease. In: Harding A, Deufel T, Chamberlain S (Editors), Hereditary ataxias. Vol. 61. New York: Raven Press; 1993. p 139-153.
7. Kieling C, Prestes PR, Saraiva-Pereira ML, Jardim LB. Survival estimates for patients with Machado-Joseph disease (SCA3). Clin Genet 2007; 72: 543-545.
8. Yamada S, Nishimiya J, Nakajima T, Taketazu F. Linear high intensity area along the medial margin of the internal segment of the globus pallidus in Machado-Joseph disease patients. J Neurol Neurosurg Psychiatry 2005; 76: 573-575.
9. Yuasa T, Ohama E, Harayama H, Yamada M, Kawase Y, Wakabayashi M, et al. Joseph’s disease: clinical and pathological studies in a Japanese family. Ann Neurol 1986; 19: 152-157.
10. Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, et al. Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol 1996; 39: 490-499.
11. Iwabuchi K, Tsuchiya K, Uchihara T, Yagishita S. Autosomal dominant spinocerebellar degenerations. Clinical, pathological, and genetic correlations. Rev Neurol 1999; 155: 255-270.
12. Cecchin CR, Pires AP, Rieder CR, Monte TL, Silveira I, Carvalho T, et al. Depressive symptoms in Machado-Joseph disease (SCA3) patients and their relatives. Community Genet 2007; 10: 19-26.
13. Riberto M, Miyazaki MH, Jucá SSH, Sakamoto H, Pinto PPN, Battistella LR. Validação da versão brasileira da medida de independência funcional. Acta Fisiatr 2004; 11: 72-76.
14. Granger CV, Hamilton BB, Keith RA, Zielezny M, Sherwin FS. Advances in functional assessment for rehabilitation. In: Anonymous, Topics in geriatric rehabilitation. Rockville: Aspen; 1986.
15. Mahoney FI, Barthel DW. Functional evaluation: the Barthel index. Md State Med J 1965; 14: 61-65.
16. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23: 56-62.
17. Fleck MP, Louzada S, Xavier M, Chachamovich E, Vieira G, Santos L, et al. [Application of the Portuguese version of the abbreviated instrument of quality life WHOQOL-bref]. Rev Saúde Pública 2000; 34: 178-183.
18. Development of the World Health Organization WHOQOL-BREF quality of life assessment. The WHOQOL Group. Psychol Med 1998; 28: 551-558.
19. Kieling C, Rieder C, Silva ACF, Saute JA, Cecchin CR, Monte TL, et al. A neurological examination score for the assessment of spinocerebellar ataxia 3 (SCA3). Eur J Neurol 2008; 15: 371-376.
20. Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006; 66: 1717-1720.
21. Foster M. Theoretical frameworks. In: Turner A, Foster M, Johnson S (Editors), Occupational therapy and physical dysfunction. 5th edn. Amsterdam: Elsevier; 2002.
22. Martins S, Coutinho P, Silveira I, Giunti P, Jardim LB, Calafell F, et al. Cis-acting factors promoting the CAG intergenerational instability in Machado-Joseph disease. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 439-446.
23. Perez-Avila I, Fernandez-Vieitez JA, Martinez-Gongora E, Ochoa-Mastrapa R, Velazquez-Manresa MG. [Effects of a physical training program on quantitative neurological indices in mild stage type 2 spinocerebellar ataxia patients]. Rev Neurol 2004; 39: 907-910.
24. Paneque HM, Reynaldo AR, Velazquez PL, Santos FN, Miranda HE, Real PN, et al. [Type 2 spinocerebellar ataxia: an experience in psychological rehabilitation]. Rev Neurol 2001; 33: 1001-1005.
25. Rub U, Seidel K, Ozerden I, Gierga K, Brunt ER, Schols L, et al. Consistent affection of the central somatosensory system in spinocerebellar ataxia type 2 and type 3 and its significance for clinical symptoms and rehabilitative therapy. Brain Res Rev 2007; 53: 235-249.
26. Monte TL, Rieder CR, Tort AB, Rockenback I, Pereira ML, Silveira I, et al. Use of fluoxetine for treatment of Machado-Joseph disease: an open-label study. Acta Neurol Scand 2003; 107: 207-210.
Acknowledgments
We are grateful to all patients who participated in this study. Research supported by CNPq (#302035/2004-4), FAPERGS, and FIPE-Hospital de Clínicas de Porto Alegre.
Address for correspondence: L.B. Jardim, Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Rua Ramiro Barcelos, 2350, 90035-903 Porto Alegre, RS, Brasil. E-mail: ljardim@hcpa.ufrgs.br
Received August 11, 2009. Accepted March 2, 2010. Available online April 16, 2010. Published June 11, 2010.
The Brazilian Journal of Medical and Biological Research is partially financed by
- 1. Takiyama Y, Nishizawa M, Tanaka H, Kawashima S, Sakamoto H, Karube Y, et al. The gene for Machado-Joseph disease maps to human chromosome 14q. Nat Genet 1993; 4: 300-304.
- 2. Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 1994; 8: 221-228.
- 3. Bauer PO, Kotliarova SE, Matoska V, Musova Z, Hedvicakova P, Boday A, et al. Fluorescent multiplex PCR - fast method for autosomal dominant spinocerebellar ataxias screening. Genetika 2005; 41: 830-837.
- 4. Prestes PR, Saraiva-Pereira ML, Silveira I, Sequeiros J, Jardim LB. Machado-Joseph disease enhances genetic fitness: a comparison between affected and unaffected women and between MJD and the general population. Ann Hum Genet 2008; 72: 57-64.
- 5. Jardim LB, Pereira ML, Silveira I, Ferro A, Sequeiros J, Giugliani R. Machado-Joseph disease in South Brazil: clinical and molecular characterization of kindreds. Acta Neurol Scand 2001; 104: 224-231.
- 6. Sequeiros J, Coutinho P. Epidemiology and clinical aspects of Machado-Joseph disease. In: Harding A, Deufel T, Chamberlain S (Editors), Hereditary ataxias. Vol. 61. New York: Raven Press; 1993. p 139-153.
- 7. Kieling C, Prestes PR, Saraiva-Pereira ML, Jardim LB. Survival estimates for patients with Machado-Joseph disease (SCA3). Clin Genet 2007; 72: 543-545.
- 8. Yamada S, Nishimiya J, Nakajima T, Taketazu F. Linear high intensity area along the medial margin of the internal segment of the globus pallidus in Machado-Joseph disease patients. J Neurol Neurosurg Psychiatry 2005; 76: 573-575.
- 9. Yuasa T, Ohama E, Harayama H, Yamada M, Kawase Y, Wakabayashi M, et al. Joseph’s disease: clinical and pathological studies in a Japanese family. Ann Neurol 1986; 19: 152-157.
- 10. Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, et al. Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol 1996; 39: 490-499.
- 11. Iwabuchi K, Tsuchiya K, Uchihara T, Yagishita S. Autosomal dominant spinocerebellar degenerations. Clinical, pathological, and genetic correlations. Rev Neurol 1999; 155: 255-270.
- 12. Cecchin CR, Pires AP, Rieder CR, Monte TL, Silveira I, Carvalho T, et al. Depressive symptoms in Machado-Joseph disease (SCA3) patients and their relatives. Community Genet 2007; 10: 19-26.
- 13. Riberto M, Miyazaki MH, Jucá SSH, Sakamoto H, Pinto PPN, Battistella LR. Validação da versão brasileira da medida de independência funcional. Acta Fisiatr 2004; 11: 72-76.
- 14. Granger CV, Hamilton BB, Keith RA, Zielezny M, Sherwin FS. Advances in functional assessment for rehabilitation. In: Anonymous, Topics in geriatric rehabilitation. Rockville: Aspen; 1986.
- 15. Mahoney FI, Barthel DW. Functional evaluation: the Barthel index. Md State Med J 1965; 14: 61-65.
- 16. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23: 56-62.
- 17. Fleck MP, Louzada S, Xavier M, Chachamovich E, Vieira G, Santos L, et al. [Application of the Portuguese version of the abbreviated instrument of quality life WHOQOL-bref]. Rev Saúde Pública 2000; 34: 178-183.
- 18. Development of the World Health Organization WHOQOL-BREF quality of life assessment. The WHOQOL Group. Psychol Med 1998; 28: 551-558.
- 19. Kieling C, Rieder C, Silva ACF, Saute JA, Cecchin CR, Monte TL, et al. A neurological examination score for the assessment of spinocerebellar ataxia 3 (SCA3). Eur J Neurol 2008; 15: 371-376.
- 20. Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006; 66: 1717-1720.
- 21. Foster M. Theoretical frameworks. In: Turner A, Foster M, Johnson S (Editors), Occupational therapy and physical dysfunction. 5th edn. Amsterdam: Elsevier; 2002.
- 22. Martins S, Coutinho P, Silveira I, Giunti P, Jardim LB, Calafell F, et al. Cis-acting factors promoting the CAG intergenerational instability in Machado-Joseph disease. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 439-446.
- 23. Perez-Avila I, Fernandez-Vieitez JA, Martinez-Gongora E, Ochoa-Mastrapa R, Velazquez-Manresa MG. [Effects of a physical training program on quantitative neurological indices in mild stage type 2 spinocerebellar ataxia patients]. Rev Neurol 2004; 39: 907-910.
- 24. Paneque HM, Reynaldo AR, Velazquez PL, Santos FN, Miranda HE, Real PN, et al. [Type 2 spinocerebellar ataxia: an experience in psychological rehabilitation]. Rev Neurol 2001; 33: 1001-1005.
- 25. Rub U, Seidel K, Ozerden I, Gierga K, Brunt ER, Schols L, et al. Consistent affection of the central somatosensory system in spinocerebellar ataxia type 2 and type 3 and its significance for clinical symptoms and rehabilitative therapy. Brain Res Rev 2007; 53: 235-249.
- 26. Monte TL, Rieder CR, Tort AB, Rockenback I, Pereira ML, Silveira I, et al. Use of fluoxetine for treatment of Machado-Joseph disease: an open-label study. Acta Neurol Scand 2003; 107: 207-210.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
16 Apr 2010 -
Date of issue
June 2010
History
-
Received
11 Apr 2009 -
Accepted
02 Mar 2010