Abstracts
A dissolution test for tablets containing 100 mg of lamotrigine was developed and validated. The dissolution test was applied to compare the dissolution profile of Neural® with the reference product Lamictal®. The analysis procedure was carried out using a simple ultraviolet method at 267 nm. After the determination of solubility and sink conditions, the parameters selected were paddles at 50 rpm, 900 mL of 0.01 M hydrochloric acid, and 30 minutes duration (single point). This method was validated for specificity, linearity, accuracy, precision and robustness. Lamotrigine stability was also evaluated in dissolution medium.
Lamotrigine; Dissolution test; Medicines
A finalidade deste estudo foi desenvolver e validar um método de dissolução para o fármaco lamotrigina na forma farmacêutica comprimido. Este método também foi utilizado para comparar o perfil de dissolução entre o Neural® e o produto de referência Lamictal®. O procedimento analítico foi realizado utilizando-se espectrofotometria de absorção no ultravioleta (267 nm) como forma de quantificação do fármaco. Após a determinação da solubilidade e das condições sink, os parâmetros selecionados foram: pás (50 rpm), 900 mL de ácido clorídrico 0.01 M e o tempo de 30 minutos (único ponto). Este método foi validado através da especificidade, linearidade, exatidão, precisão e robustez. A estabilidade da lamotrigina também foi avaliada no meio de dissolução.
Lamotrigina; Teste de dissolução; Medicamentos
ARTICLE
Development of a dissolution test for lamotrigine in tablet form using an ultraviolet method
Magda Targa Martins* * Correspondence: M. T. Martins. Departamento de Farmácia, Faculdade de Farmácia, Universidade Federal do Rio Grande do Sul. Av. Ipiranga, 2752 Lab. 402 - 906010-000 - Porto Alegre - RS, Brazil E-mail: magdatm@terra.com.br ; Clésio Soldatelli Paim; Martin Steppe
Departamento de Farmácia, Faculdade de Farmácia, Universidade Federal do Rio Grande do Sul
ABSTRACT
A dissolution test for tablets containing 100 mg of lamotrigine was developed and validated. The dissolution test was applied to compare the dissolution profile of Neural® with the reference product Lamictal®. The analysis procedure was carried out using a simple ultraviolet method at 267 nm. After the determination of solubility and sink conditions, the parameters selected were paddles at 50 rpm, 900 mL of 0.01 M hydrochloric acid, and 30 minutes duration (single point). This method was validated for specificity, linearity, accuracy, precision and robustness. Lamotrigine stability was also evaluated in dissolution medium.
Uniterms: Lamotrigine. Dissolution test/validation. Medicines/quality control.
RESUMO
A finalidade deste estudo foi desenvolver e validar um método de dissolução para o fármaco lamotrigina na forma farmacêutica comprimido. Este método também foi utilizado para comparar o perfil de dissolução entre o Neural® e o produto de referência Lamictal®. O procedimento analítico foi realizado utilizando-se espectrofotometria de absorção no ultravioleta (267 nm) como forma de quantificação do fármaco. Após a determinação da solubilidade e das condições sink, os parâmetros selecionados foram: pás (50 rpm), 900 mL de ácido clorídrico 0.01 M e o tempo de 30 minutos (único ponto). Este método foi validado através da especificidade, linearidade, exatidão, precisão e robustez. A estabilidade da lamotrigina também foi avaliada no meio de dissolução.
Unitermos: Lamotrigina. Teste de dissolução/validação. Medicamentos/controle de qualidade.
INTRODUCTION
Drug absorption after oral administration depends on the release of the drug's active substance from the drug formulation, whereas clinical effect depends on the amount of the drug that reaches the site of action (Ansel, 2000; Shargel et al., 2005). Dissolution tests are an important tool in various steps of drug production and quality control. The dissolution tests for immediate release, solid, oral dosage forms such as tablets, are used to assess lot-to-lot quality of a drug product; to guide development of new drug formulations and ensure consistent product quality and performance after certain changes such as alterations in formulation and the manufacturing process (FDA, 1997). Lamotrigine (The Merck Index, 2001; Figure 1), 3,5-diamino-6-(2,3-dichorophenyl)-1,2,4-triazine, [84057-84-1], C9H7Cl2N5, mol. wt. 256.09, is chemically unrelated with other antiepileptic drugs in current use. This drug is useful in the treatment of partial and tonic-clonic generalized seizures resistant to other anticonvulsant drugs (Cordioli, 2000). It acts through stabilizing pre-synaptic membranes by blocking voltage-dependent sodium channels. The drug has also been associated with preventing aspartate and glutamate release (Kliemann, 2004). The efficacy of lamotrigine as an add-on therapy has been well established in adults and children (Ben-Menachen, 2000) and was recently approved as monotherapy (Cheng et al., 2005).
Although there are many studies describing the determination of lamotrigine in biological fluids (Cocciglio et al., 1991; Sallustio, Morris, 1997; Matar et al., 1998; Ashton et al., 1999; Botinger et al., 1999; Vidal et al., 1999; Angelis Stofordis et al., 1999; Barbosa, Midio, 2000; Torra et al., 2000; Croci et al., 2001; Castel-Branco et al., 2001; Patil, Bodhankar, 2005; Cheng et al., 2005; Kuldeep, Subash, 2005), and pharmaceutical formulation (Emani et al., 2006; Yousef, Taha, 2007) by several methods besides a dissolution test using HPLC method (Sripalakit et al., 2008), no dissolution test for this pharmaceutical solid dosage form has yet been described in any pharmacopoeia. Therefore, this paper describes the development and validation of a dissolution test for lamotrigine in tablets using a simple, fast and inexpensive ultraviolet method, and performs a comparative evaluation of the dissolution profiles of two different formulations. The development and validation were carried out in compliance with the United States Pharmacopeia (USP 31, 2008).
MATERIAL AND METHODS
Experimental
Instrumentation
The dissolution tests were performed on a multi-bath (n=7) dissolution test system Sotax AT7 (Basel, Switzerland), in accordance with the United States Pharmacopeia (USP 31, 2008) general methods.
An UV-VIS Recording Spectrophotometer UV-160A (Shimadzu, Kyoto, Japan) at 267 nm, using 1.0 cm quartz cells and SPECTRA MANAGER software, was used for all absorbance measurements to quantify the samples of lamotrigine dissolved in the dissolution medium.
The Digimed potentiometer, model DM-20 (São Paulo, Brazil), was used to determine the pH of solutions.
The ultrasonic-bath UCS 5000 (Unique, São Paulo, Brazil) was used for deaeration of the media and preparation of solutions.
The filter used for sample filtration was a Framex® quantitative filter of 10 mm. A 0.45µm, 13 mm, nylon membrane Millex® (Millipore, São Paulo, Brazil) was used for mobile phase filtration in stability and solubility determination by the HPLC method.
A model LC-1200 series liquid chromatograph (Agilent, Santa Clara, CA, United States) equipped with G1311A Quaternary pump, ALS G1329A auto sampler, TCC G1316A Thermostat Column Compartment, DAD G1315B photodiode-array detector, SCL-10Avp system controller and Chemstation manager system software, was employed for lamotrigine stability and solubility determination.
Chemicals and reagents
The lamotrigine reference substance (99.41%) was obtained from Cristália Produtos Químicos Farmacêuticos LTDA (São Paulo, Brazil). The pharmaceutical formulations (Product A and Product B) containing lamotrigine were obtained commercially.
Product A - (reference product, Lamictal®) - labeled as containing 100 mg of lamotrigine and the following inactive ingredients (lactose, microcrystalline cellulose, povidone, sodium starch glycolate, ferric oxide (yellow) and magnesium stearate).
Product B - (Neural®) - labeled as containing 100 mg of lamotrigine and the following inactive ingredients (mannitol, pregelatinized starch, dibasic calcium phosphate, corn starch, sodium starch glycolate, magnesium stearate, and tartrazine yellow colorant).
All of the inactive ingredients were obtained from different local distributors. Purified water was obtained using a Millipore® system (São Paulo, Brazil). HPLC grade triethylamine and orthophosphoric acid were purchased from Merck® (Darmstadt, Germany). Hydrochloric acid (HCl), sodium hydroxide and sodium acetate were purchased from Quimex® (São Paulo, Brazil). Monobasic potassium phosphate was obtained from Synth® (São Paulo, Brazil). Glacial acetic acid was obtained from Nuclear® (Brazil). The 0.01 and 0.1 M HCl, 0.05 M sodium acetate buffer (pH 4.7), 0.05 M monobasic potassium phosphate buffer (pH 5.8 and 6.8) were prepared according to the USP (USP 31 2008).
Chromatographic conditions
A method using HPLC with UV detection at 279 nm was previously developed and validated in our laboratory for a wide range of parameters including specificity, linearity, precision, accuracy and robustness. Chromatographic separation was achieved on an ACE® RP-18 octadecyl silane column (150 mm x 4.6 mm i.d., particle size 5 µm). The mobile phase was composed of triethylamine solution 0.3% pH 4.0 (adjusted with 10% orthophosphoric acid) and methanol (62:38, v/v). The mobile phase flow rate was 1.0 mL.min-1 with isocratic elution and was filtered before use through a 0.45 µm membrane filter Millex® (Millipore, São Paulo, Brazil) and degassed. The injection volume was 20 µL and the column temperature was maintained at 25 ºC. The results obtained demonstrated that the method is specific (there were no peaks by placebo solution interfering in the lamotrigine retention time), linear (ranging from 10.0 to 80.0 mg.mL-1 for analysis of variance (ANOVA) and correlation coefficient 0.9999), precise (with RSD values of less than 2%), accurate by recovery test (98.20% to 100.46%) and robust (the changes in operational parameters did not cause significant differences in the performance of chromatographic system).
Solubility determination and sink conditions
Lamotrigine sink conditions were applied and determined in different media: purified water, 0.01 M and 0.1 M hydrochloric acid, 0.05 M acetate buffer (pH 4.7) and 0.05 M phosphate buffer (pH 5.8 and 6.8). Vessels (n=2) containing 10.0 ml of medium were pre-heated to 37ºC ± 0.5ºC before adding an excess of lamotrigine (10.0 mg). The solutions were maintained in the bath and stirred with a vortex at intervals of 10 minutes. Aliquots of 10.0 mL were removed from each vessel after 1 and 2 hours and filtered through a Millipore® 0.45 µm membrane filter. Aliquots of 1.0 mL of the filtered solution were transferred to 25.0 mL volumetric flasks, diluted with mobile phase and injected into the HPLC. Standard solution of lamotrigine was prepared with 40 µg.mL-1 and used to determine the amount of lamotrigine dissolved in different media.
Dissolution test conditions
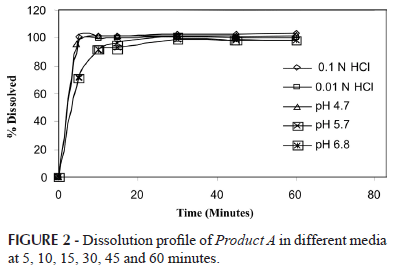
Dissolution testing of tablets was performed with Product A in accordance with the USP (USP 31, 2008) to define the dissolution test conditions. This was performed using paddles (USP apparatus 2) at a stirring speed of 50 rpm and 900 mL of the different dissolution media pre-heated to 37 ºC ± 0.5 ºC. Manual sampling aliquots of 10.0 ml were withdrawn at 5, 10, 15, 30, 45 and 60 minutes.
After these preliminary studies, the dissolution test was conducted using the conditions described above and the medium selected was 0.01 M hydrochloric acid maintained at 37 ± 0.5 ºC. The profile was evaluated and a single point at 30 minutes was selected. After 30 minutes, aliquots of 10.0 mL were withdrawn from each vessel and filtered using a Framex® quantitative filter.
The sample solutions were prepared using the filtered aliquots taken from the dissolution vessels. The 4.0 mL aliquots were transferred to 20.0 ml volumetric flasks with 0.01 M hydrochloric acid and analyzed on a UV/VIS Spectrophotometer (267 nm).
The standard solution was prepared using an amount of lamotrigine reference substance equivalent to 11.11 mg, transferred to a 20 mL volumetric flask with the dissolution medium. The solutions were placed in an ultrasonic-bath for 15 minutes. An aliquot of 2.0 mL of this standard solution was transferred to a 50.0 mL volumetric flask and diluted with 0.01 M hydrochloric acid to give a solution containing 22.22 µg.mL-1. This solution was analyzed on a UV/VIS spectrophotometer at 267 nm.
Filtration
Filtration of the dissolution samples is usually necessary to prevent undissolved drug particles from entering the analytical sample and removes insoluble excipients that otherwise cause high background or turbidity. The filter evaluation is necessary to verify whether it can be used in the dissolution test without adsorption of the drug into the filter (USP 31, 2008). Standard and sample solutions were prepared in the dissolution medium proposed (0.01 M hydrochloric acid). The standard solution was prepared with a lamotrigine concentration of 0.11 mg.mL -1. This solution was placed for 15 minutes in an ultrasonic-bath (Solution A). Four milliliters of Solution A was diluted in a 20.0 mL volumetric flask with 0.01 M hydrochloric acid - (solution without filtration). The remaining Solution A was filtered using a Framex® quantitative filter, and an aliquot of 4.0 mL was transferred to a 20.0 mL volumetric flask and diluted with the same solvent - (filtered solution). The samples were prepared with the same concentration of 0.11 mg.mL -1 using 0.01 M hydrochloric acid. This solution was maintained for 15 minutes in an ultrasonic bath and immediately centrifuged at 3000 rpm for 10 minutes. An aliquot of 4.0 ml was transferred to a 20.0 mL volumetric flask and diluted with 0.01 M hydrochloric acid - (solution without filtration). Another solution was prepared in the same manner, but instead of undergoing centrifugation was filtered with a Framex® quantitative filter, and an aliquot of 4.0 ml was transferred to a 20.0 ml volumetric flask and diluted with the same solvent - (filtered solution). The standard solutions (filtered and without filtration) and the sample solutions (centrifuged and filtered) were analyzed using UV determination at 267 nm.
Validation
In order to demonstrate the method's suitability for use as a dissolution test, it was validated based on stability, specificity, linearity, precision, accuracy and robustness parameters (USP 31, 2008).
Stability
The standard solution stability test was performed in the dissolution medium selected (0.01 M hydrochloric acid) at 37 ºC ± 0.5 ºC for 2 hours and at room temperature for 24 hours. The chromatograms obtained by the HPLC method were evaluated and compared with a freshly prepared standard solution (peak area and degradation product formation).
Specificity
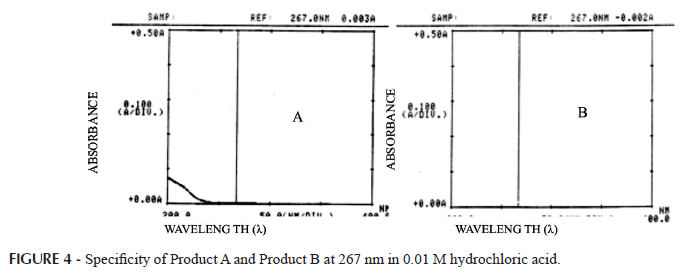
Placebo samples of both commercial products were prepared in their usual compositions. The placebo samples were transferred to different vessels (n=3) with 900 mL of dissolution medium at 37 ºC ± 0.5 ºC and stirred for 2 h at 150 rpm using a paddle (USP apparatus 2). Aliquots of these solutions were filtered through a FRAMEX® quantitative filter and analyzed by the UV method (267 nm).
Linearity
A stock solution was prepared containing 100 µg mL-1 of lamotrigine using 0.01 M hydrochloric acid as solvent. Aliquots of this solution were transferred to volumetric flasks to obtain final concentrations of 5, 10, 15, 20, 25, 30 and 40 µg.mL-1. Each solution was prepared in triplicate. The linearity was evaluated by linear regression analysis, which was calculated by the least squares regression method and analysis of variance (ANOVA).
Precision
The evaluation of inter-day precision (intermediate precision) of the dissolution test was performed on two different days by different analysts. The repeatability was evaluated on the same day for intra-day precision in six vessels used for the dissolution test. The relative standard deviation (RSD) from the results was calculated.
Accuracy
A lamotrigine reference stock solution (2.0 mg.mL-1) was prepared in 0.01 M hydrochloric acid), using an ultrasonic-bath for 60 minutes. A recovery study was conducted by adding known amounts of the lamotrigine stock solution to the dissolution vessels containing the placebo solution (10, 20 and 100%) of the nominal assay (100 mg). Each concentration was prepared in duplicate and analyzed by the UV method at 267 nm.
Robustness
This was evaluated by comparing the dissolution medium with and without using the ultrasonic bath. Medium deaeration was used to eliminate air bubbles before the dissolution test. The results from dissolution samples in nondeaerated medium and deaerated medium had to be compared to determine whether deaeration was necessary (USP 31, 2008).
RESULTS AND DISCUSSIONS
Drug solubility is an important physical-chemical property to be considered when selecting the dissolution medium and can be determined by using an excess of the drug in the medium followed by stirring, filtration and quantification of the dissolved drug (USP 31, 2008). This is the property that most affects the drug dissolution velocity ( Abdou, 1989). Lamotrigine presents limited solubility in water (0.17 mg.mL-1) at 25 ºC, and its solubility decreases in proportion to pH increase in, since this drug is a weak basis and is not ionized at a pH greater than its pKa (5.7). Dissolution characteristics must be tested in the physiological pH range (1.2 to 6.8) for immediate drug release, and can be considered fast drug dissolution when no less than 85% of the drug amount is dissolved within 15 minutes (USP 31, 2008; Yu et al., 2002). Sink condition is defined to be no less than three times the necessary volume to obtain a saturated drug solution (Marques, Brown, 2002). Thissink condition was reached for the media in which lamotrigine solubility was higher than 333.33 µg.mL-1,i.e. three times the nominal dose in 900 mL. This occurred in the following media: 0.01 M hydrochloric acid, 0.1 M hydrochloric acid, acetate buffer pH 4.7, phosphate buffer pH 5.8 and after 2 hours in phosphate buffer pH 6.8. Sink conditions were not reached in purified water or in phosphate buffer pH 6.8, 1 hour (Table I). In HCl media (0.1 M and 0.01 M) however, the solubility must have been higher since all the drug dissolved. Water is not an ideal dissolution medium for several reasons. First, because the variability of the quality of the water from different sources, and second, the variability of the pH from day to day and also during the run, where this depends on the active substance and excipients (FDA, 1997; USP 31, 2008).
The dissolution profile at six points (5, 10, 15, 30, 45 and 60 minutes) of Product A in the evaluated media showed that lamotrigine dissolved more than 85% in 15 minutes (Figure 2). Thus, for a very rapidly dissolving drug, a simple one point dissolution test is all that may be needed and for immediate-release dosage forms, the duration of the procedure is typically 30 to 60 minutes (Amidon et al., 1995; USP 31, 2008). This was expected from the results obtained in the solubility test. The choice of the medium was 0.01 M hydrochloric acid due to its good solubility, accessibility, low cost and the fact that it is a typical dissolution medium (Marques, Brown, 2002).
The initial parameters for filtration and solution stability must be established prior to the completion of any dissolution samples (Fortunato, 2005). The results between the solutions (filtered or centrifuged) must be very similar for acceptance of the filter (Skoug, Freeman, 1996). The evaluation of the filter demonstrated that the FRAMEX® can be used safely during the dissolution test, since there was no significant drug adsorption. The difference in values between the solutions was less than 2%. In order to evaluate lamotrigine stability in 0.01 M hydrochloric acid, chromatograms were obtained. The results (Figure 3) showed that the solutions remained stable and that no degradation peaks formed. According to the literature (USP 31, 2008), the acceptable range for solution stability is within 98-102% of the initial value. The values obtained were less than 1% in both conditions evaluated (99.90% for solution maintained at 37 ºC ± 0.5 ºC for 2h, and 99.25% at room temperature for 24 h) compared with a freshly prepared standard solution. Therefore, it was possible to guarantee the integrity of the drug during all the analysis time.
The specificity analysis of placebo solutions revealed that the UV method (267 nm) suffered no interference from the formulation for either of the products evaluated (A and B), demonstrating this method to be specific (Figure 4). Since there were not inactive ingredients interfering with the wavelength selected (267 nm), UV was the method selected for lamotrigine quantification (Fortunato, 2005). Generally, the simplest assay method to develop utilizes UV/VIS spectrophotometry. This method is frequently used in quality control of pharmaceutical products due to the potential of the great majority of the drugs that absorb energy at these wavelengths. Lamotrigine presents a structure with aromatic rings, allowing absorption in the UV region. The UV method requires no expensive or complex equipment, and needs no toxic solvents. (Bakshi, 2002). The preferred method of analysis is spectrophotometric determination because results can be obtained faster, the analysis is simpler and fewer solvents are used, making this valuable in routine analysis (USP 31, 2008; Skoug, Freeman, 1996).
To assess linearity, three calibration curves for lamotrigine were constructed, plotting concentration (µg mL-1) versus absorbance. This demonstrated linearity, showing a correlation coefficient of approximately 0.9999 in the concentration range studied (4.0-40.0 µg.mL-1). The representative linear equation was y= 0.02723x + 0.00229. The data were validated by means of the analysis of variance (ANOVA), which demonstrated significant linear regression and no significant linearity deviation (p<0.05) (Farmacopéia Brasileira, 1988).
The precision results of the dissolution method were evaluated by analyzing repeatability and intermediate precision. The assay evidenced a low RSD ranging from 0.70 to 1.52 for intra-day precision and 1.23 for inter-day precision. RSD values lower than 2% indicated the good precision of this method.
Degree of accuracy expresses the concordance between the accepted value and the value found. The measured recovery is typically 95-105% (USP 31, 2008). Mean recovery obtained was in the range of 98.46 to 103.59%, which indicated good accuracy of the method.
In the evaluation of method robustness, the presence of possible air bubbles in the dissolution medium did not interfere in the dissolution profile of lamotrigine in tablets. Based on the results obtained, and considering the percentage of dissolved drug (102.85% in deaerated medium and 101.57% in nondeaerated medium), the use of an ultra-sonic bath is not necessary because the difference between the results was not significant (1.28%).
Lamotrigine BCS (Biopharmaceutics Classification System) is class I, with high solubility and high permeability, and these products need not be subjected to a profile comparison if they can be shown to release 85% or more of the active drug substance within 15 minutes (Anderson, 2008). Consequently, it is not necessary to calculate f1 (measure of the percentage error between two curves over all time points) and f2 (logarithmic transformation of the sum squared error of differences between the test and reference over all time points), since they have lost their discriminative power (Brasil, 2004). Acceptance criteria for the amount of active ingredient dissolved, expressed as a percentage of the labeled content (Q), are in the range of 75% to 80% dissolved ingredient (USP 31, 2008). In this study on lamotrigine, we can propose an acceptance criteria of 80% ingredient dissolved in 30 minutes. The results obtained for the mean amount of lamotrigine dissolved for both products in 30 minutes were (Product A - 101.41% and Product B - 98.37%) and the comparison of their profiles is shown in Figure 5. Amindon et al. (1995) suggested that products with high solubility such as lamotrigine can be considered bioequivalent if they allow 85% of drug release within 15 minutes. The products studied (A and B) were in agreement with this specification, but in vivo studies need to be carried out to guarantee the bioequivalence of the products. The amount of dissolved drug can be influenced by many factors including the manufacturing process, formulation, humidity and substances properties (Skoug et al., 1996). The comparison of the dissolution profiles allowed differences between the formulations to be observed.
CONCLUSIONS
The dissolution test developed and validated for lamotrigine was considered satisfactory. The method has the advantages of being very easy, fast and simple. The method was carefully assessed to guarantee the stability of the drug during the analysis time. The method is suitable for its purpose and could be applied in routine quality control of lamotrigine in tablet formulation since there is no official monograph for this drug in the pharmacopoeias.
ACKNOWLEDGEMENTS
The authors would like to thank LCQFar and LEPCQ.
Received for publication on 22nd January 2009
Accepted for publication on 25th June 2009
- ABDOU, H. M. Dissolution, Bioavailability & Bioequivalence. Easton: Mack Publishing Company, 1989. 554p.
- AMIDON, G. L.; CRISON, J. R.; LENNERNAS, H.; SHAH, V. P. A theoretical basis for a biopharmaceutic drug classification: the correlation in vitro drug product dissolution and in vivo bioavailability. Pharm. Res., v.12, n.3, p.413-419, 1995.
- ANDERSON, D. G. Understanding the ramifications of switching among AEDs: What are the data? Adv. Stud. Med., v.8, n.7, p.229-234, 2008.
- ANGELIS-STOFORDIS, P.; MORGAN, D. J.; O'BRIEN, T. J.; VAJDA, F. J. E. Determination of lamotrigine in human plasma by high-performance liquid chromatography. J. Chromatogr. B, v.727, n.1, p.113-118, 1999.
- ANSEL, H. C.; ALLEN Jr., L.; POPOVICH, N. Desenvolvimento de novos fármacos e processos de aprovação São Paulo: Premier, 2000. 568 p.
- ASHTON, D. S.; RAY, D. A.; VALKO, K. Detection of principal routine indicative impurity in lamotrigine. Int. J. Pharm., v.189, n.2, p.241-248, 1999.
- BAKSHI, M.; SARANJIT, S. Development of validated stability-indicating assay methods - critical review. J. Pharm. Biomed. Anal., v.28, n.6, p.1011-1040, 2002.
- BARBOSA, N. R.; MIDIO, A. F. Validated high-performance liquid chromatography method for the determination of lamotrigine in human plasma. J. Chromatogr. B, v.741, n.2, p.289-293, 2000.
- BEN-MENACHEN, E. New antiepileptic drugs and non-pharmacological treatments. Curr. Opin. Neurol., v.13, n.2, p.165-170, 2000.
- BOTTIGER, Y.; SVENSSON, J. O.; STAHLE, L. Lamotrigine drug interactions in a TDM material. Ther. Drug Monit, v.21, n.2, p.171-174, 1999.
- BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RE nº310, 1º de setembro de 2004. Determina a publicação da guia para a realização do estudo e elaboração do relatório de equivalência farmacêutica e perfil de dissolução. Diário Oficial da República Federativa do Brasil, Brasília, 2004. Seção?, p.1-3.
- BUDAVARI. The MERCK INDEX. An encyclopedia of chemicals, drugs, and biologicals. Whitehouse Station: Merck & Co. Inc., 2001. 2564 p.
- CASTEL-BRANCO, M. M.; ALMEIDA, A. M.; FALCÃO, A. C.; MACEDO, T. A.; CERAMONA, M. M.; LOPEZ, F. G. Lamotrigine analysis in blood and brain by high-performance liquid chromatography. J. Chromatogr. B, v.755, n.1-2, p.119-121, 2001.
- CHENG, C. L.; CHOU, C. H.; HU, O. Y. Determination of lamotrigine in small volumes of plasma by high-performance liquid chromatography. J. Chromatogr. B., v.817, n.2, p.199-206, 2005.
- CORDIOLI, A. V. Psicofármacos Porto Alegre: Artes Médicas, 2000. 556 p.
- COCIGLIO, M.; ALRIC, R.; BOUVIER, O. J. Performance analysis of a reversed phase liquid chromatography assay of lamotrigine in plasma using solvent demixing extraction. J.Biomed Applic, v.110, n.1-2, p.269-276, 1991.
- CROCI, D.; SALMAGGI, A.; GRAZIE, U. New high-performance liquid chromatography. Ther. Drug Monit., v.23, n.6, p.665-668, 2001.
- EMANI, J.; GHASSAMI, N.; AHMADI, F. Development and validation of a new HPLC method for determination of lamotrigine and related compounds in tablet formulations. J. Pharm. Biomed. Anal., v.40, n.4, p.999-1005, 2006.
- FARMACOPÉIA BRASILEIRA. 4.ed. Rio de Janeiro: Atheneu, 1988. 1320 p.
- FDA. Guidance for industry: Dissolution testing of immediate release solid oral dosage forms. U.S. Department of health and human services, Food and Drug Administration, Center for drug evaluation and research (CDER). Rockville: FDA, 1997. p.1-17.
- FORTUNATO, D. Dissolution method development for immediate release solid oral dosage forms. Dissol. Tech., v.12, n.3, p.12-15, 2005.
- KLIEMANN, A. D. F.; MONTE, T. L. Antiepilépticos. In: FUCHS, F. D; WANNMACHER, L.; FERREIRA, M. B. C., Eds. Farmacologia clínica- Fundamentos da terapêutica racional. 3.ed. Rio de Janeiro: Guanabara Koogan, 2004. p.535-552.
- KULDEEP, M. P.; SUBHASH, L. B. HPTLC determination of lamotrigine in serum. J. Chromatogr. B., v.823, n.2, p.152-157, 2005.
- MARQUES, M. R. C.; BROWN, W. Desenvolvimento e validação de métodos de dissolução para formas farmacêuticas sólidas orais. Analytica, v.1, n.1, p.48-51, 2002.
- MATAR, K. M.; NICHOLS, A. S.; BAWAZIR, A. S.; AL-HASSAN, M. I.; TEKLE, A. A. A rapid liquid chromatography method for the determination of lamotrigine in plasma. J. Pharm. Biomed. Anal, v.17, n.3, p.525-531, 1998.
- PATIL, K. M.; BODHANKAR, S. L. Simultaneous determination of lamotrigine, phenobarbitone, carbamazepine and phenytoin in human serum by high-performance liquid chromatography. J. Pharm. Biomed. Anal, v.39, n.1-2, p.181-186, 2005.
- SALLUSTIO, B. C.; MORRIS, R. G. High performance liquid chromatography quantitation of plasma lamotrigine concentrations: application measuring trough concentrations in patients with epilepsy. Ther. Drug Monit, v.19, n.6, p.688-693, 1997.
- SHARGEL, L.; WU-PONG, S.; YU, A. B. C. Applied biopharmaceutics & pharmacokinetics. New York: McGraw-Hill, p.267-269, 2005.
- SKOUG, J. W.; HALSTEAD, D. L.; THEIS, D. L.; FREEMAN, J.; FAGAM, D.; RHORS, B. R. Roteiro para desenvolvimento e validação do teste de dissolução em formas farmacêuticas sólidas para uso oral. Pharm. Tech., v.20, n.5, p.58-72, 1996.
- SRIPALAKIT, P.; SRICHAYA, A.; KANDEE, R. Development and validation of a HPLC method for a dissolution test of lamotrigine tablets and its application to drug quality control studies. J. Liq. Chromatogr. Relat. Technol, v.31, n.6, p.926-939, 2008.
- THE UNITED STATES Pharmacopeia. 30.ed. Rockville: United States Pharmacopeial Convention, 2008. p.3796-3800.
- TORRA, M.; ROODAMILANS, M.; ARROYO, S.; CRBELLA, J. Optimised procedure for lamotrigine analysis in serum by high performance liquid chromatography without interferences from other frequently coadministered anticonvulsants. Ther. Drug Monit, v.22, n.5, p.621-625, 2000.
- VIDAL, E.; PASCUAL, C.; POU, L. Determination of lamotrigine in human serum by high-performance liquid chromatography. J. Chromatogr. B, v.736, n.1, p.295-298, 1999.
- YOUSEEF, N. F.; TAHA, E. A. Development and validation of spectrophotometric, TLC and HPLC methods for determination of lamotrigine in presence of its impurity. Chem. Pharm. Bull, v.55, n.4, p.541-545, 2007.
- YU, L. X.; AMIDON, G. L.; POLLI, J. E.; ZHAO, H.; METHA, M. U.; CONNER, D. P.; SHAH, V. P.; LESKO, L. J.; CHEN, M.; LEE, V. H. L.; HUSSAIN, A. S. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm. Res, v.19, n.7, p.921-925, 2002.
Publication Dates
-
Publication in this collection
17 Aug 2010 -
Date of issue
June 2010
History
-
Received
22 Jan 2009 -
Accepted
25 June 2009