Abstracts
PURPOSE: To study and compare the appearance of hairs from patients with Chédiak-Higashi and Griscelli-Prunieras syndromes under light and polarized light microscopy. METHOD: Hairs from 2 Chédiak-Higashi and 2 Griscelli-Prunieras patients were obtained and examined under normal and polarized light microscopy. RESULTS: Under light microscopy, hairs from Chédiak-Higashi patients presented evenly distributed, regular melanin granules, larger than those seen in normal hairs. Under polarized light microscopy, shafts exhibited a bright and polychromatic refringence appearance. In contrast, hair from Griscelli-Prunieras patients, under light microscopy, exhibited bigger and irregular melanin granules, distributed mainly near the medulla. Under polarized light microscopy, shafts appeared monotonously white. CONCLUSION: Light microscopic examination of hair shafts of patients with Chédiak-Higashi or Griscelli-Prunieras syndrome reveals subtle differences that are useful in identifying both disorders, but not in distinguishing between them. We provide evidence that polarized light microscopy of hair shafts, an approach that has not been previously described, aids in differentiating between these syndromes. We propose hair study by polarized light microscopy as a helpful complementary diagnostic method for differential diagnosis between CHS and GPS, especially when the more sophisticated molecular studies are not available.
Chédiak-Higashi; Griscelli-Prunieras; Silvery hair; Polarized light microscopy
OBJETIVO: Estudar e comparar o aspecto dos cabelos de portadores das síndromes de Chédiak-Higashi e Griscelli-Prunieras, tanto na microscopia óptica convencional quanto com luz polarizada. MÉTODO: Cabelos de dois doentes portadores da síndrome de Chédiak-Higashi e de dois portadores da síndrome de Griscelli-Prunieras foram obtidos e estudados tanto à microscopia convencional quanto com luz polarizada. RESULTADOS: Na microscopia óptica convencional, os cabelos dos doentes portadores da síndrome de Chédiak-Higashi mostraram grânulos de melanina regulares, com distribuição homogênea e de maior tamanho em comparação aos presentes no cabelo normal. À microscopia de luz polarizada notou-se aspecto brilhante e refringência policromática. Diferentemente, os cabelos dos doentes portadores da síndrome de Griscelli-Prunieras apresentaram à microscopia convencional, grânulos de melanina irregulares e maiores do que os presentes no cabelo normal e os presentes nos cabelos dos doentes portadores da síndrome de Chédiak-Higashi, preferencialmente próximos à medula das hastes pilosas. À microscopia de luz polarizada apresentaram aspecto monotonamente esbranquiçado. CONCLUSÃO: O exame dos cabelos pela microscopia convencional nas síndromes de Chédiak-Higashi e Griscelli-Prunieras revela diferenças sutis no reconhecimento dessas doenças. No presente trabalho apresentamos evidência de que o exame das hastes pilosas com microscopia de luz polarizada - não descrito previamente - contribui na diferenciação de ambas doenças sugerindo que esse seja um método diagnóstico útil na distinção entre as síndromes de Chédiak-Higashi e Griscelli Prunieras, especialmente nos casos em que estudos moleculares mais sofisticados não estejam disponíveis.
Cabelo Prateado; Chédiak-Higashi; Griscelli-Prunieras; Microscopia óptica de luz polarizada
ORIGINAL RESEARCH
Polarized light microscopy of hair shafts aids in the differential diagnosis of Chédiak-Higashi and Griscelli-Prunieras syndromes
Contribuição do estudo dos cabelos com microscopia de luz polarizada ao diagnóstico diferencial das síndromes de Chédiak-Higashi and Grisceli-Prunieras
Neusa Y.S.Valente, Maria Cecilia M.R. Machado, Paula Boggio, Ana Cristina F. Alves, Fabiane N. Bergonse, Erasmo Casella , Dewton Moraes Vasconcelos, Anete S. Grumach and Zilda N.P. de Oliveira
Dermatology Department / Pediatrics Department /Outpatient Unit and Laboratory of Medical Investigation in Allergy and Immunology (LIM-56) / Hospital das Clinicas, Faculty of Medicine, University of São Paulo (USP), São Paulo, Brazil. Email: paulaboggio@terra.com.br
ABSTRACT
PURPOSE: To study and compare the appearance of hairs from patients with Chédiak-Higashi and Griscelli-Prunieras syndromes under light and polarized light microscopy.
METHOD: Hairs from 2 Chédiak-Higashi and 2 Griscelli-Prunieras patients were obtained and examined under normal and polarized light microscopy.
RESULTS: Under light microscopy, hairs from Chédiak-Higashi patients presented evenly distributed, regular melanin granules, larger than those seen in normal hairs. Under polarized light microscopy, shafts exhibited a bright and polychromatic refringence appearance. In contrast, hair from Griscelli-Prunieras patients, under light microscopy, exhibited bigger and irregular melanin granules, distributed mainly near the medulla. Under polarized light microscopy, shafts appeared monotonously white.
CONCLUSION: Light microscopic examination of hair shafts of patients with Chédiak-Higashi or Griscelli-Prunieras syndrome reveals subtle differences that are useful in identifying both disorders, but not in distinguishing between them. We provide evidence that polarized light microscopy of hair shafts, an approach that has not been previously described, aids in differentiating between these syndromes. We propose hair study by polarized light microscopy as a helpful complementary diagnostic method for differential diagnosis between CHS and GPS, especially when the more sophisticated molecular studies are not available.
Key words: Chédiak-Higashi; Griscelli-Prunieras; Silvery hair; Polarized light microscopy.
RESUMO
OBJETIVO: Estudar e comparar o aspecto dos cabelos de portadores das síndromes de Chédiak-Higashi e Griscelli-Prunieras, tanto na microscopia óptica convencional quanto com luz polarizada.
MÉTODO: Cabelos de dois doentes portadores da síndrome de Chédiak-Higashi e de dois portadores da síndrome de Griscelli-Prunieras foram obtidos e estudados tanto à microscopia convencional quanto com luz polarizada.
RESULTADOS: Na microscopia óptica convencional, os cabelos dos doentes portadores da síndrome de Chédiak-Higashi mostraram grânulos de melanina regulares, com distribuição homogênea e de maior tamanho em comparação aos presentes no cabelo normal. À microscopia de luz polarizada notou-se aspecto brilhante e refringência policromática. Diferentemente, os cabelos dos doentes portadores da síndrome de Griscelli-Prunieras apresentaram à microscopia convencional, grânulos de melanina irregulares e maiores do que os presentes no cabelo normal e os presentes nos cabelos dos doentes portadores da síndrome de Chédiak-Higashi, preferencialmente próximos à medula das hastes pilosas. À microscopia de luz polarizada apresentaram aspecto monotonamente esbranquiçado.
CONCLUSÃO: O exame dos cabelos pela microscopia convencional nas síndromes de Chédiak-Higashi e Griscelli-Prunieras revela diferenças sutis no reconhecimento dessas doenças. No presente trabalho apresentamos evidência de que o exame das hastes pilosas com microscopia de luz polarizada - não descrito previamente - contribui na diferenciação de ambas doenças sugerindo que esse seja um método diagnóstico útil na distinção entre as síndromes de Chédiak-Higashi e Griscelli Prunieras, especialmente nos casos em que estudos moleculares mais sofisticados não estejam disponíveis.
Unitermos: Cabelo Prateado; Chédiak-Higashi; Griscelli-Prunieras; Microscopia óptica de luz polarizada.
INTRODUCTION
Chédiak-Higashi syndrome (CHS) is a very rare genetic disorder, inherited in an autosomal recessive mode. It is clinically characterized by mild pigmentary dilution, silvery-blond hair, severe phagocytic immune deficiency, bleeding tendencies, recurrent pyogenic infection, progressive neurological defects, and early death from a lymphoma-like hemophagocytic syndrome.1-5
In 1978, Griscelli and Prunieras reported an autosomal recessive syndrome having clinical features and a course that resembled CHS, leading to the term, Chédiak-Higashi-like syndrome.6,7 Nowadays, it is a well characterized entity known as Griscelli-Prunieras syndrome (GPS).
The correct diagnosis and differentiation between CHS and GPS requires light microscopic examination (and, when possible, electron microscopic study) of skin and hair shafts, as well as immunological and peripheral blood smear evaluations. Determinations for specific genetic defects for each syndrome are currently available.
The aim of this paper is to describe the polarized microscopic aspect of hair shafts from patients with CHS and with GPS. To the best of our knowledge, there are no previous reports in the English literature focusing on hair appearance under polarized light microscopy in these diseases.
METHODS
Case reports
Case 1:
A 20-month-old black girl, born to consanguineous (first-degree cousins) parents, presented since birth with generalized skin hypopigmentation and ash-gray hair. During follow-up, sun-exposed areas of skin have become darker, although flexures and the trunk have remained hypochromic. At the age of 8 months, she developed recurrent bacterial pneumonias. Under light microscopic skin examination, many large melanin granules were detected throughout the epidermis. Blood smear evaluation showed the presence of giant peroxidase-positive granules within granulocytes (Fig. 1), confirming the CHS diagnosis.
Case 2:
A 5-year-old white female, born to nonconsanguineous parents, presented with generalized skin hypopigmentation and silvery hair since birth. By the age of 4 months, her skin became darker on the face and light-brown with a grayish hue on the extremities, sparing flexural areas. Otherwise, she was in good health. Light microscopic skin study of a hyperpigmented area on the forearm showed many large melanin granules inside melanocytes and keratinocytes. Peripheral blood smear examination, in which large peroxidase-positive granules inside neutrophils and lymphocytes were found, led to the CHS diagnosis.
Immunological data showed hypergammaglobulinemia and reduced stimulated chemotaxis in both CHS patients (Table I).
Case 3:
A 7-year-old white boy, born to first-cousin parents, was referred to our clinic due to silvery hair and a history of recurrent respiratory infections. The physical and neurological examinations were normal. Light microscopic skin examination (on skin of normal appearance) revealed dense hyperpigmentation of basal melanocytes, little pigment in the surrounding epidermis, and absence of giant melanin granules. The peripheral blood smear investigation was normal. Immunological data showed an impaired stimulated NBT (nitroblue tetrazolium) test (Table I). Clinical and laboratory data suggested a GPS diagnosis.
Case 4:
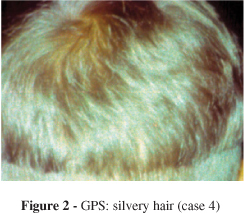
A 4-year-old white male, born to a nonconsanguineous couple, was admitted due to a progressive sleepy state. On physical examination, silvery-gray hair was noted (Fig. 2), and we were asked to evaluate him. A skin biopsy showed dense hyperpigmented basal melanocytes with sparse pigmentation in adjacent keratinocytes. Immunological studies showed low plasma levels of IgA and IgG for the patient's age (Table I). The peripheral blood smear was normal. He evolved with severe motor neurological deterioration, recurrent infections, and a progressive hepatosplenomegaly associated with myeloid metaplasia, leading to introduction of immunosuppressive therapy. With these clinical, skin, and hair microscopic findings, a GPS diagnosis was proposed.
At least 20 hair shafts from each of these 4 patients (2 females with CHS and 2 males with GPS) were collected and prepared on a glass slide with Entellan resin and a glass coverslip. Light microscopy examination, with and without polarization, was then performed.
RESULTS
Light microscopic hair examination
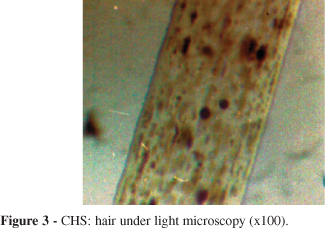
CHS patients: shafts showed evenly distributed melanin granules of regular diameter that were bigger than those seen in normal hair (Fig. 3).
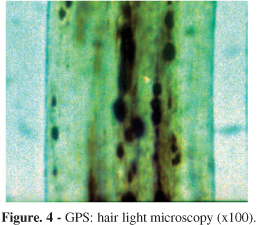
GPS patients: shafts presented large unevenly melanin granules, mostly located in vicinity of the medullar zone. They were larger than those described for normal hair as well as those seen in CHS hair (Fig. 4).
Polarized light microscopic hair examination
CHS patients: bright shafts with different individual colors were detected, resembling normal white hairs (Fig. 5).
GPS patients: shafts looked bright with a monotonously whitish appearance (Fig. 6).
DISCUSSION
The main signs of dermatologic interest in CHS and GPS are generalized skin hypopigmentation at birth with tanning capacity after sun exposure, hairs with a unique silvery sheen, and more rarely, pigmentary dilution of the iris. This phenotype has been described as "partial albinism".8,9 Albinism comprises a group of hereditary disorders in which defective genes participate in the melanin synthesis pathway; while the pigmentary alterations in CHS and GPS depend on impaired transference of melanosomes from melanocytes to keratinocytes due to mutations in some genes regulating intracellular organelle transport.10,11 Because of these molecular differences between albinism and CHS/GPS, the two latter entities are preferably designated as "silvery hair syndromes".10-12
Chédiak-Higashi syndrome (OMIM #214500) is a very rare systemic disorder, inherited as an autosomal recessive trait.13 Parental consanguinity is frequently reported in affected patients,12,14 but this was only confirmed in case 1 of our series. The defective CHS gene, called CHS1/LYST, maps to chromosome 1q42-43.5 It participates in regulation of lysosomal trafficking, and when altered, causes an aberrant fusion of diverse intracellular organelles, giving rise to the abnormally giant granules seen in several tissues that constitutes the hallmark of CHS.4,5,9,15,16
Dermatologic manifestations in CHS range from mild-to-severe pigmentary dilution of skin, hair, and eyes. The fair skin of these patients usually takes a slate-gray hue, as seen in our cases 1 and 2. It appears to be less pigmented when compared to other family members and displays an increased susceptibility to sunburn.16 Hyperpigmentation of sun-exposed areas, as occurred in both of our CHS patients, has been previously described, but a satisfactory explanation for this rare observation is still absent.16,17 An unusual gray or silvery metallic sheen of scalp hair (present in the 2 patients reported here), eyelashes, and eyebrows is reported as the most conspicuous finding.13 The iris ranges from blue to brown.13 Strabismus, nystagmus, and photophobia are common, but visual acuity is usually normal.3,5
Immunological dysfunction manifests as early, severe, and recurrent infectious episodes; mainly involving the skin, the upper respiratory tract, and the lungs. The most common causative organisms isolated are Staphylococcus aureus, Streptococcus pyogenes and S. pneumoniae.3,5,7,15,18-20 The main immunologic defects consist of impaired chemotaxis of granulocytes, together with defective intracellular killing, diminished antibody-dependent cytotoxicity, and defective natural killer cell function.4,5,9,13,21 A history of several bacterial pneumonias and chemotaxis in the lower limit were established for case 1.
Patients who live long enough may develop progressive neurological deterioration, resembling spinocerebellar degeneration.4,5,22 Near adolescence, most patients enter the so-called "accelerated phase," characterized by pancytopenia, hepatosplenomegaly, and lymphohistiocytic infiltrates into major organs. It has a ruinous prognosis, and death generally occurs from infection or bone marrow failure.13,21 Currently, the only curative treatment is bone marrow transplantation.9 To date, neither of our patients had developed neurological impairment nor entered the accelerated phase.
Griscelli-Prunieras syndrome (GPS), also known as "partial albinism with immunodeficiency," is an autosomal recessive disorder. Parental consanguinity was established in one of our two cases. The identification of specific genetic defects underlying GPS is the basis of the current classification into 3 types. In both GPS type 1 (GPS1-OMIM #214450), also known (and preferentially designated) as Elejalde syndrome (ES-OMIM #256710), and GPS type 2 (GPS2-OMIM #607624), the mutated genes map to chromosome 15q21.17,25 Recently, GPS type 3 (GPS3) has been recognized as being determined by a mutation at a different band (2.37) of the same chromosome 15.23 Products of the altered genes in GPS participate in melanosome transference from melanocytes to keratinocytes.
All the GPS subgroups share the same phenotype of pigmentary dilution. Classical findings resemble those of CHS, i.e., silvery-gray hair, pale skin with persistent capacity for tanning when sun-exposed, and ocular alterations secondary to pigment diminution.7,22,24 In both GPS patients reported here, the most evident dermatologic finding was the presence of silvery hair.
Although having a similar pigmentary phenotype, patients with GPS have different prognoses depending on the subtype. Patients with GPS2 present a history of severe infections caused by immunologic defects (absence of delayed-type cutaneous hypersensitivity and impaired natural killer cell function).22 Hypogammaglobulinemia has also been reported.7,22 Interestingly, low levels of both serum IgA and IgG were detected in patient 4. In advanced disease, patients develop an acute phase of uncontrolled lymphocyte and macrophage activation (hemophagocytic disorder). Based on clinical and laboratory findings, we concluded that both of our GPS cases correspond to GPS2. In contrast, patients with GPS1 never evolve to accelerated phases, but present progressive and more severe neurological disturbance that results from the limited expression of MYO5A (the defective gene in GPS1) in the central nervous system.25 Finally, clinical expression for patients with GPS3 will be limited to the pigmentary alterations already described. The prognosis for GPS1 and GPS2 is poor. For patients with GPS2, treatment is the same as for CHS patients.7,22 Currently, there is no specific treatment for GPS1, which always has a fatal outcome.22,25 In contrast, the prognosis for patients with GPS3 is good, and these patients do not require any treatment.
For cases in which establishing a differential diagnosis between CHS and GPS is difficult, chromosome analysis detecting the specific causative mutation would be more appropriate. Nevertheless, such evaluation is not easily available, especially in developing countries.
Polarized light microscopy is used to examine doubly refractile materials, such as urate crystals, amyloid, cholesterol esters, silica, and certain foreign bodies.26 Keratin also has doubly refractile properties. Therefore, normal hair shafts can be seen under polarized light microscopy as more or less bright bodies, depending on their baseline hair color.
In our experience, findings from polarized light microscopy obtained when analyzing hairs from patients with CHS or GPS complement data regarding the size and distribution of the hair melanin granules that are found under light microscopic examinations. Polarized light microscopy reveals the whitish, monotonous aspect of hairs of patients with GPS, contrasting clearly with their polychromatic appearance in CHS patients. This may be considered a differential elementnot previously reportedfor distinguishing between these syndromes (CHS and GPS), especially when data from light microscopic hair examination is inconclusive and/or molecular studies are not available. The reasons for this variation as revealed by polarized light microscopy are not known, and we have not found a convincing explanation.
Received for publication on March 29, 2006
Accepted for publication on April 20, 2006
- 1. Stegmaier OC, Schneider LA. Chédiak-Higashi syndrome. Arch Dermatol. 1965;91:1-9.
- 2. Blume RS, Wolff SM. The Chédiak-Higashi syndrome: studies in four patients and a review of the literature. Medicine. 1972;4:247-78.
- 3. Barak Y, Nir E. Chédiak-Higashi syndrome. Am J Pediatr Hematol Oncol. 1987;9:42-55.
- 4. Shiflett SL, Kaplan J, Mcvey Ward D. Chédiak-Higashi syndrome: a rare disorder of lysosomes and lysosome related organelles. Pigment Cell Res. 2002;15:251-7.
- 5. Paller AS. Chédiak-Higashi syndrome. In: Textbook of pediatric dermatology. Harper J, Orange A, Prose N, editors. Oxford: Blackwell Science; 2000: 1681-2.
- 6. Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, Prunieras M. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- 7. Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency: Griscelli syndrome: report of a case and review of the literature. J Am Acad Dermatol. 1998;38:295-300.
- 8. Ortonne JP. Chédiak-Higashi syndrome. In: Dermatology. Bolognia JL, Jorizzo JL, Rapini RP, editors. Philadelphia: Mosby, Elsevier Limited; 2003: 960.
- 9. Ganschow R, Grabhorn E, Lemke J, Lepler L. Cardiac failure in an infant with Chédiak-Higashi syndrome: a hypothesis of the effect of diadenosine polyphosphates. Pediatr Allergy Immunol. 2002;13:307-10.
- 10. Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, de la Luz-Orozco-Covarrubias M, Tamayo L, Ruiz-Maldonado R. Elejalde syndromea melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-6.
- 11. Lambert J, Vancoillie G, Naeyaert JM. Elejalde syndrome revisited. Arch Dermatol. 2000;136:120.
- 12. Cahali JB, Fernandez SA, Oliveira ZN, Machado MC, Valente NS, Sotto MN. Elejalde syndrome: report of a case and review of the literature. Pediat Dermatol. 2004;21:479-82.
- 13. Arbiser JL. Genetic immunodeficiencies: cutaneous manifestations and recent progress. J Am Acad Dermatol. 1995;33:82-9.
- 14. Grumach AS, Duarte AJ, Bellinati-Pires R, Pastorino AC, Jacob CM, Diogo CL, et al. Brazilian report on primary immunodeficiencies in children: 166 cases studied over a follow-up time of 15 years. J Clin Immunol. 1997;17:340- 5.
- 15. Barton LM, Roberts P, Trantou V, Haworth C, Kelsey H, Blamires T. Chédiak-Higashi syndrome. Br J Haematol. 2004;125:2.
- 16. Al-Khenaizan S. Hyperpigmentation in Chédiak-Higashi syndrome. J Am Acad Dermatol. 2003;49:S244- 6.
- 17. Fukai K, Ishii M, Kadoya A, Chanoki M, Hamada T. Chédiak-Higashi syndrome: report of a case and review of the Japanese literature. J Dermatol. 1993;20:231-7.
- 18. Pritz RA. Chédiak-Higashi syndrome. In: Primary immunodeficiency diseases: a molecular and genetic approach, 1st ed. Ochs HD, Smith CIE, Puck JM, editors. New York: Oxford University Press; 1999:389-96.
- 19. Carnide EM, Jacob CM, Pastorino AC, Bellinati-Pires R, Costa MB, Grumach AS. Chédiak-Higashi syndrome: presentation of seven cases. Sao Paulo Med J. 1998;116:1873-8.
- 20. Bellinati-Pires R, Salgado MM, Joazeiro PP, Carneiro-Sampaio MM. Delayed phagocytosis and bacterial killing in Chédiak-Higashi syndrome neutrophils detected by a fluorochrome assay. Ultrastructural aspects. Mem Inst Oswaldo Cruz. 1992;87:575-81.
- 21. Demirkiran O, Utku T, Urkmez S, Dikmen Y. Chédiak-Higashi syndrome in the intensive care unit. Pediatr Anesthesia. 2004;14:685-8.
- 22. Scheinfeld NS. Syndromic albinism: a review of genetics and phenotypes. Dermatol Online J. 2003;9:5.
- 23. Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-60.
- 24. Westbroek W, Lambert J, De Schepper S, Kleta R, Van Den Bossche K, Seabra MC, et al. Rab27b is up-regulated in human Griscelli syndrome type II melanocytes and linked to the actin cytoskeleton via exon F-myosin Va transcripts. Pigment Cell Res. 2004;17:498-505.
- 25. Klein C, Philippe N, Le Deist F, Fraitag S, Prost C, Durandy A, et al. Partial albinism with immunodeficiency (Griscelli syndrome). J Pediatr. 1994;125:886-95.
- 26. Lever WF and Schaumburg-Lever G. Laboratory methods. In: Histopathology of the skin, 6th ed. Lever WF and Schaumburg-Lever G, editors. Philadelphia: JB Lippincott Company; 1983: p. 45.
Publication Dates
-
Publication in this collection
16 Aug 2006 -
Date of issue
Aug 2006
History
-
Received
29 Mar 2006 -
Accepted
20 Apr 2006