MINI-REVIEW
Cytogenetic and molecular contributions to the study of mental retardation
Juan C. Llerena Jr. and José Carlos Cabral de Almeida
Centro de Genética Médica, Instituto Fernandes Figueira, FIOCRUZ, Av. Rui Barbosa, 716, 22250-000 Rio de Janeiro, RJ, Brasil. E-mail: llerena@iff.fiocruz.br and Unidade de Citogenética Humana, Instituto de Biofísica CCFº/UFRJ, Brasil. Send correspondence to J.C.L.Jr.
A remarkable improvement in the diagnosis and a noticeable advance in the understanding of the pathogenesis of disorders with mental retardation (MR) have been obtained through well-documented clinical observation of rare diseases in humans, coupled with the application of a variety of newly developed molecular genetic techniques and the discovery of new genetic processes in the cause of diseases.
A total of 827 diseases associated with MR are listed under the McKusick's Mendelian Inheritance in Man Catalog (OMIM, 1998). This is a comprehensive reference for monogenic MR causes, classified under autosomal dominant (201 - 24%), autosomal recessive (425 - 51%), X-linked (135 - 16%), mitochondrial inheritance (6 - 0.6%), and multiple entries since 1994 (50 - 6%).
In this brief review of causes of MR we have selected five main groups, as follows:
· Contiguous gene syndromes
· Cryptic chromosomal aberrations
· X-inactivation abnormalities
· Genomic imprinting errors
· Mitochondrial mutations
Other genetic mechanisms leading to MR such as expanded trinucleotide-related diseases (Mandel, 1997; Reddy and Housman, 1997) have also been extensively investigated. However, they will not be discussed here.
Contiguous gene syndromes (CGS)
Outstandingly accurate cytogenetic characterization of several CGS has been accomplished with the optimization of in situ hybridization techniques which, with the availability of a vastly increased number of new probes, has narrowed the FISH (fluorescent in situ hybridization) investigation to a 2-Mb resolution.
The Smith-Magenis syndrome, del(17)(p11.2) (Figure 1), is a good example of how the phenotype of this frequently underdiagnosed CGS has been expanded by the definite diagnosis of the micro-cytogenetic aberration by FISH (Greenberg et al., 1996). Furthermore, chromosomal constitutional structural microdeletion mosaics, which are recognized as being very rare and frequently non-convincingly diagnosed by conventional or even high resolution cytogenetic techniques, can be, at present, properly excluded or confirmed (Juyal et al., 1995, 1996). The FISH technique, besides permitting a correct diagnosis, allows retrospective case investigation of syndromes/sequences where a heterogeneous phenotype is expected (the Velo-Cardio-Facial syndrome, del(22)(q12), is a good example) (Goldberg et al., 1993; Swilllen et al., 1997).
A 16-year-old boy with typical Smith-Magenis syndrome. FISH investigation using specific 17p11.2 probe revealed deleted chromosome 17 (arrow).
Another important contribution of FISH to the clinical field of MR resides in its application to genetic coun seling in such conditions as, for instance, the Miller-Dieker syndrome [del(17)(p13.3)], for which an unusually high frequency of cryptic balanced rearrangements involving region 17p13.3 in either parent has been reported (Pilz et al., 1995; Yokoyama et al., 1997). Particular multiple congenital anomaly syndromes with MR attributed to a recessive pattern of inheritance may be unmasked by FISH and shown to be due to a subtle parental translocation undetected by conventional cytogenetic techniques, including high resolution preparations, as recently demonstrated by Herens et al. (1997) in the Lambotte syndrome. Furthermore, the application of nuc ish (interphase/nuclear in situ hybridization) (ISCN, 1995) to archival pathology slides has rescued chromosome aberration diagnosis on previously undiagnosed fetal wastage material (Figure 2) or malformed patients, permitting, in several cases, an adequate genetic counseling.
Cell suspension from a first trimester miscarriage investigated by FISH using chromosome 1 a-centromeric probe. Nuc ish with three signals indicated a presumably triploid conceptus.
In Table I, recognized syndromes characterized by partial monosomy associated with MR which may be due to haploinsufficiency genes are listed. Of the many chromosome deletion syndromes identified, positional cloning has so far led to the isolation of only a few genes or candidate genes (OMIM, 1998). These genes fall into a number of different categories such as transcriptional regulators (PAX6,GLI-3,ZNF-141,TUPLE-1), receptors and signal transduction molecules (RET,LIS1), and ribosomal protein homologues (Fisher and Scambler, 1994).
Human partial monosomy syndromes with MR which may be due to haploinsufficiency (based on Fisher and Scambler, 1994; OMIM, 1998).
The correlation of phenotype-genotype and haploinsufficiency genes, coupled with biochemical techniques and molecular embryology investigation, has signaled a new field of research concerning dosage-sensitive developmental pathways both in humans and in animal models (Reeves et al., 1995; Aalfs et al., 1997).
Cryptic chromosomal aberrations
Hidden chromosomal abnormalities have been continuously unraveled by the implementation of new FISH probes, especially those involved in complex chromosomal rearrangements (CCR) and/or in apparently balanced autosomal translocations associated with an abnormal phenotype with MR. Figure 3 exemplifies a CCR G-banded karyotype of a young non-dysmorphic boy referred for investigation because of MR. A CCR involving four chromosomes (1q, 6q, 7p, 15q) was detected. Using specific painting and cosmid probes an unusual abnormality, not previously delineated by conventional techniques, was characterized as a composition of different segments of 1p and 6q, alternately interspersed in the derivative chromosome 7 short arm. The 15q derivative chromosome present in this complex rearrangement was shown to be a paracentric inversion and a further Y chromosome aberration was identified in the rearrangement. At least 9 breakpoints have to be invoked to explain the unusual rearrangement observed in the proband's chromosomes. The karyotype and the observed mental retardation are probably related, and a clinical screening protocol is being followed in the search for malignancies, considering that the majority of the chromosomal breakpoints resides within proto-oncogene loci (Cabral de Almeida et al., 1997).
(a) G-banded karyotype showing a complex chromosome rearrangement involving chromosomes 1p;6q;7p;15q. FISH investigation: (b) whole chromosome painting probes for chromosomes 1 and 6 revealed a complex structural rearrangement of the der(7) (see text); (c) a paracentric inversion of chromosome 15 using specific 15q probes was characterized; (d) translocation of telomeric 7p to the distal Yq region was further identified.
Cases of abnormal phenotypes associated with apparently balanced reciprocal translocations have been revised through the narrowing of boundaries at the breakpoint sites by specific molecular probes (Crolla et al., 1996). Figure 4 illustrates deletions of PAX6 and WT1 genes and an apparently balanced 11p13;13q33 translocation in a young girl with isolated aniridia, without MR. The order of probes used in this investigation revealed that apart from PAX6 gene deletion, the WT1 gene was also missing, resulting in a high risk for Wilms tumor, which however was not detected. The most telomeric 11p probe used, cosmid F1238, mapping to region 11p14.2, was preserved, indicating that the putative MR gene locus of the WAGR complex may be located distal to this site since our patient did not have MR.
(a) Two partial metaphases showing an apparently reciprocal 11p13;q33 translocation; (b) whole chromosome painting probes for chromosomes 11 and 13 confirm the reciprocal translocation. WT1 (c) and PAX6 (d) genes were deleted within the patient's chromosomal aberration.
X-inactivation abnormalities
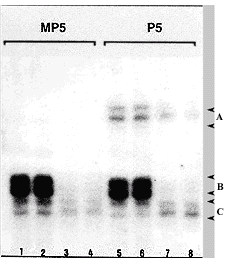
Along with the known occurrence of X-linked diseases with MR (XLMR) in heterozygote female patients and well- documented correlation between XLMR and X skewed inactivation for Lesch-Nyhan syndrome (Ogasawara et al., 1989), pyruvate dehydrogenase deficiency (Brown et al., 1994) and fra(X) (Willard, 1996), exciting new data have been put forward relating the X-chromosome inactivation process and diseases associated with MR. The pioneer investigation by Sarto and Therman's group (Sarto et al., 1987) of the X-inactivation pattern of patients with a 49,XXXXY or XXXXX karyotype has shown that a highly asynchronous replication pattern of the late-replicating X chromosomes was observed in their experiments and could be correlated to the impairment of cognitive development. This asynchrony could maintain extra X-chromosome regions active through critical periods of the cell cycle leading to a more severe phenotype, with special emphasis on MR in the 49,X cases compared to other X aneuploidies (Linden et al., 1995). Recently, we investigated the origin of the extra haploid set of 5 individuals carrying a 49,XXXXY or 49,XXXXX karyotype. All cases turned out to be maternal in origin. Molecular studies using the androgen receptor methylation pattern of three informative mothers showed a skewed pattern of X-chromosome inactivation in all of them (Figure 5). It has been shown that 3 to 9% of normal females in the general population present a skewed pattern of X-inactivation (Harris et al., 1992; Pegoraro et al., 1997). The above observations seem to indicate a possible causative relationship between maternal skewed X inactivation and X-polysomies.
Parental origin (MP5 - mother) and androgen receptor (AR) methylation experiments in a 49,XXXXX patient (P5). Lanes 1,2,5 and 6 - total DNA amplified by PCR for exon 1 from AR gene; lanes 3,4,7 and 8 show PCR amplified DNA after CfoI digestion. Note maternal origin of the extra X chromosomes and maternal skewed inactivation pattern in lanes 3 and 4 (alelle B < alelle C).
Another very important group of MR patients associated with the X-inactivation process is represented by mentally retarded Turner syndrome patients with a mos 45,X/46,X,der(X) karyotype (Migeon et al., 1994). We investigated three patients with typical Turner syndrome phenotype, MR and 45,X karyotype in blood and skin cultures. Buccal smear nuc ish using X-centromeric probe revealed the presence of a second X centromere in a few cells of all three patients. These findings are probably related to the severe MR observed in the patient and could be a consequence of the absence or non-functional XIST RNA that inappropriately activates the second derivative X chromosome, most often represented by small fragments, resulting in functional disomy of genes in the X-pericentromeric region (Willard, 1996) .
Genomic imprinting errors
Much has been learned about the mechanisms and functions of genomic imprinting by the Prader-Willi (PW) and Angelman (AS) syndromes (Glenn et al., 1997). The loss of imprinted gene expression within chromosome 15q11-q13 leads to different phenotypes, depending on the parental origin of the aberration. These different phenotypes are a consequence of a paternal chromosomal deletion or "functional" genetic monosomy in PW patients and maternal in influence AS patients.
Four types of molecular mechanisms have been distinguished in the causation of PW and AS syndromes, as summarized in Table II. It should be emphasized that some types are observed more frequently in one disease than in the other. UPD is more frequently associated with PW syndrome because it is generally secondary to a mitotic event (loss of a paternal chromosome) following a trisomic zygote due to maternal meiosis I nondisjunction. In AS, since paternal non-disjuntion is much less frequent, UPD is rare and due to either secondary to paternal meiosis II nondisjunction, or postzygotic event of maternal 15 duplication (Nicholls et al., 1992; Feil and Kelsey, 1997).
Molecular types of Prader-Willi (PW) and Angelman (AS) syndrome patients (Glenn et al., 1997).
1 Uniparental disomy.
2 Imprinting center.
More recently, an imprinting center which controls initial resetting of the parental imprint in the germline for all imprinted gene expression over a 1.5-2.5-Mb region within chromosome 15q11-q13 has been identified through microdeletions upstream of the SNRPN gene in both PW and AS patients with a 50% risk of recurrence in sibs (Glenn et al., 1997). Rare families with AS sibs, attributed to a gene mutation of the putative AS gene, favors an autosomal dominant type of inheritance with an obvious similar high recurrence risk (Saitoh et al., 1997).
The mechanisms of imprinted gene expression are not yet fully understood, but it is clear that DNA methylation and chromatin structure play an important role in both somatic cell expression and inheritance of the imprint (Kass et al., 1997; Feil and Kelsey, 1997).
Mitochondrial mutations
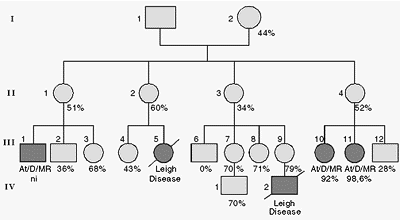
The mitochondrial genome shows strict maternal inheritance and the vast majority of copies are identical at birth (homoplasmy). Occasionally, pathogenic mutations can occur inside the mitochondrial DNA (mtDNA). When this heteroplasmic mtDNA is present, it can lead to a variety of clinical features, resulting predominantly in a series of neuro-muscular disorders, with other systems possibly also being involved (Johns, 1995; Lithtowlers et al., 1997). Figure 6 illustrates one pedigree investigated by our group in collaboration with Brian Robinson (Toronto, Canada) (Llerena Jr. et al., 1997) where an 8993 T®C mutation was detected in several members of the family. The clinical features of the affected patients revealed typical cerebral pathological findings of Leigh syndrome in the deceased propositus and his deceased cousin (III.5 and IV.2, respectively) and MR/ataxia/dysarthria (clinically resembling neurogenic muscular weakness, ataxia and retinitis pigmentosa - NARP) in two other cousins (III.10 and III.11). The affected sibs had 92 and 98.6% mtDNA heteroplasmy, respectively. The clinical presentation of the affected individuals showed differences among them, such as variations in the mode of onset, course and progression of the disease. Their diagnosis were also performed under different clinical criteria. Virus infections seemed to have been a common denominator in the complication of the clinical evolution in all four patients with severe decline in motor, speech and mental skills. All members of the family, except one (III.6), carried the mutation in different proportions of heteroplasmy, ranging from 34 to 79%, with no apparent clinical manifestations. Serious implications for genetic counseling for these mtDNA mutations are imposed since the pattern of segregation of mtDNA mutation should be considered more as a consequence of random genetic drift (Jenuth et al., 1996) than classical Mendelian inheritance. An initial estimate of recurrence rate for this family should be considered to be high.
- Family A pedigree showing typical maternal inheritance of MtDNA 8993 T®C mutation through 4 generations. The percentages relate to the heteroplasmic mutation frequency detected in each individual investigated.
Future perspectives
Despite many recent advances in human genetics, the etiology of at least 40% of severe MR remains unknown. Many of these unexplained causes of MR may involve genetics and the research in this field has been incessant. Flint et al. (1995), using highly informative DNA markers mapped to the subtelomeric regions of all chromosomes, found 3% of cryptic deletions in a group of idiopathic MR patients. New techniques such as comparative genomic hybridization have been technically optimized and should soon be included in clinical genetic practice for the investigation of MR, as has recently been reported in patients with multiple congenital anomalies where de novo structural karyotype abnormalities have been demonstrated (Levy et al., 1997; Erdel et al., 1997). We would expect that submicroscopical chromosomal aberrations could be further detected by this powerful technique, especially in the "idiopathic" group, or even in well-recognized clinical syndromes, such as Cornelia de Lange, Schinzel Giedion, among others. In these syndromes no convincing genetic etiology has yet been established, a fact that in a way, brings us back to the prophetical paper by Schmickel in 1986 on CGS, where he proposed that Williams and Rubinstein-Taybi syndromes would turn out to be examples of microdeletion syndromes.
ACKNOWLEDGMENTS
The authors wish to acknowledge Brian Robinson (Ontario, Canada) for mtDNA mutation screening, Christine Joyce and John Crolla (Salisbury, UK) for FISH investigation on PAX3, WT1 and multiple probes employed in CCR, Denilce Sumita and Mayana Zatz (Biociências, USP, São Paulo) for AR methylation studies and the researchers Hilda Ramos, Elenice Bastos, Lucia Moraes, Vera Moura and José Humberto de Abreu from Rio de Janeiro (CGM/IFF/FIOCRUZ & UCH/IBCCFº/UFRJ) for their collaboration. This work was partially supported by FINEP, FIOCRUZ and CNPq (J.C.L. No. 300564/89-9).
(Received May 11, 1998)
- Aalfs, C.M., Fantes, J.A., Wenniger-Prick, L.J.J.M., Sluijter, S., Hennekam, R.C.M., van Heyningen, V. and Hoovers, J.M.N. (1997). Tandem duplication of 11p12-p13 in a child with borderline development delay and eye abnormalities: dose effect of the PAX6 gene product? Am. J. Med. Genet.73: 267-271.
- Brown, G.K., Otero, L.J., LeGris, M. and Brown, R.M. (1994). Pyruvate dehydrogenase deficiency. J. Med. Genet. 31: 875-879.
- Cabral de Almeida, J.C., Llerena, J.C., Santa Rosa, A.A., Joyce, C. and Crolla, J. (1997). A complex chromosomal rearrangement with at least 9 breakpoints in a mentally retarded boy characterized by FISH. Am. J. Hum. Genet. 61 (Suppl.): 120 (Abstract).
- Crolla, J.A., Cross, I., Atkey, N., Wright, M. and Oley, C.A. (1996). FISH studies in a patient with sporadic aniridia and t(7;11)(q31.2;p13). J. Med. Genet.33: 66-68.
- Erdel, M., Duba, H.-C., Verdorfer, I., Lingenhel, A., Geiger, R., Gutenberger, K.-H., Ludescher, E., Utermann, B. and Utermann, G. (1997). Comparative genomic hybridization reveals a partial de novo trisomy 6q23-qter in an infant with congenital malformations: delineation of the phenotype. Hum. Genet. 99: 596-601.
- Feil, R. and Kelsey, G. (1997). Genomic imprinting: a chromatin connection. Am. J. Hum. Genet.61: 1213-1219.
- Fisher, E. and Scambler, P. (1994). Human haploinsufficiency - one for sorrow, two for joy. Nat. Genet.7: 5-7.
- Flint, J. (1995). The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat. Genet.9: 132-139.
- Glenn, C.C., Driscoll, D.J., Yang, T.P. and Nicholls, R.D. (1997). Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol. Hum. Reprod.3: 321-332.
- Goldberg, R., Motzkin, B., Marion, R., Scambler, P.J. and Shprintzen, R.J. (1993). Velo-Cardio-Facial Syndrome: a review of 120 patients. Am. J. Med. Genet.45: 313-319.
- Greenberg, F., Lewis, R.A., Potocki, L., Glaze, D., Parke, J., Killian, J., Murphay, M.A., Williamson, D., Brown, F., Dutton, R., McCluggage, C., Friedman, E. Sulek, M. and Lupski, J.R. (1996). Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2). Am. J. Med. Genet.62: 247-254.
- Harris, A., Collins, J., Vetrie, D., Cole, C. and Bobrow, M. (1992). X inactivation as a mechanism of selection against lethal alleles: further investigation of incontinentia pigmenti and X linked lymphoproliferative disease. J. Med. Genet.29: 608-614.
- Herens, C., Jamar, M., Alvarez-Gonzalez, M.-L., Lesenenfant, S., Lombete, J., Bonnivert, J., Koulischer, L. and Verloes, A. (1997). Private multiple congenital anomaly syndromes may result from unbalanced subtle translocations: t(2q;4p) explains the Lambotte syndrome. Am. J. Med. Genet. 73: 127-131.
- ISCN (1995). An International System for Human Cytogenetic Nomenclature (Mitelman, F., ed.). S. Karger, Basel.
- Jenuth, J.P., Peterson, A.C., Fu, K. and Shoubridge, E.A. (1996). Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat. Genet.14: 146-151.
- Johns, D.R. (1995). Mitochondrial DNA and disease. N. Engl. J. Med.333: 638-644.
- Juyal, R.C., Finucane, B., Shaffer, L.G., Lupski, J.R., Greenberg, F., Scott, C.I., Baldini, A. and Patel, P.I. (1995). Apparent mosaicism for del(17)(p11.2) ruled out by fluorescence in situ hybridization in a Smith-Magenis syndrome patient. Am. J. Med. Genet.59: 406-407.
- Juyal, R.C., Kuwano, A., Kondo, I., Zara, F. Baldini, A. and Patel, P.I. (1996). Mosaicism for del(17)(p11.2p11.2) underlying the Smith-Magenis syndrome. Am. J. Med. Genet.66: 193-196.
- Kass, S.U., Pruss, D. and Wolffe, A.P. (1997). How does DNA methylation repress transcription. Trends Genet. 13: 444-449.
- Levy, B., Gershin, I.F., Desnick, R.J., Babu, A., Gelb, B.D., Hirschhorn, K. and Cotter, P.D. (1997). Characterization of a de novo unbalanced chromosome rearrangement by comparative genomic hybridization and fluorescent in situ hybridization. Cytogenet. Cell Genet. 76: 68-71.
- Lightowlers, R.N., Chinnery, P.F., Turnbull, D.M. and Howell, N. (1997). Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet. 13: 450-455.
- Linden, M.G., Bender, B.G. and Robinson, A. (1995). Sex chromosome tetrasomy and pentasomy. Pediatrics96: 672-682.
- Llerena Jr., J.C., Cabral de Almeida, J.C., Serapiăo, M. and Robinson, B. (1997). MtDNA 8993T-C mutation in a family with Leigh syndrome and MR/Ataxia/Dysarthria. Am. J. Hum. Genet.61 (Suppl.): 314 (Abstract).
- Mandel, J.-L. (1997). Breaking the rule of three. Nature386: 767-769.
- Migeon, B.R., Luo, S., Jani, M. and Jeppesen, P. (1994). The severe phenotype of females with tiny ring X chromosomes is associated with inability of these chromosomes to undergo X inactivation. Am. J. Hum. Genet.565: 497-504.
- Nicholls, R.D., Shashidhar Pai, D., Gottlieb, W. and Cantú, E.S. (1992). Paternal uniparental disomy of chromosome 15 in a child with Angelman syndrome. Ann. Neurol.32: 512-518.
- Ogasawara, N., Stout, J.T., Goto, H., Sonta, S., Matsumoto, A. and Caskey, C.T. (1989). Molecular analysis of a female Lesch-Nyhan patient. J. Clin. Invest.84: 1024-1027.
- OMIM (1998). Online Mendelian Inheritance in Man, OMIM (TM) (http://www.ncbi.nlm.nih.gov/OMIM/.). Center for Medical Genetics, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD).
- Pegoraro, E., Whitaker, J., Mowery-Rushton, P., Surti, U., Lanasa, M. and Hoffman, E.P. (1997). Familial skewed X inactivation: a molecular trait associated with high spontaneous-abortion rate maps to Xq28. Am. J. Hum. Genet.61: 160-170
- Pilz, D.T., Dalton, A., Long, A., Jaspan, T., Maltby, E.L. and Quarrell, O.W.J. (1995). Detecting deletions in the critical region for lissencephaly on 17p13.3 using fluorescent in situ hybridisation and a PCR assay identifying a dinucleotide repeat polymorphism. J. Med. Genet. 32: 275-278.
- Reddy, P.S. and Housman, D.E. (1997). The complex pathology of trinucleotide repeats. Curr. Opin. Cell. Biol.9: 364-372.
- Reeves, R.H., Irving, N.G., Moran, T.H., Wohn, A., Kitt, C., Sisodia, S.S., Schmidt, C., Bronson, R.T. and Davisson, M.T. (1995). A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 11: 177-184.
- Saitoh, S., Buiting, K., Cassidy, S.B., Conroy, J.M., Driscoll, D.J., Gabriel, J.M., Gillessen-Kaesbach, G., Glenn, C.C.,Greenswag, L.R., Horsdhemke, B., Kondo, I., Kuwajima, K., Niikawa, N., Rogan, P.K., Schwartz, S., Seip, J., Williams, C.A. and Nicholls, R.D. (1997). Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation. Am. J. Med. Genet.68: 195-206.
- Sarto, G., Otto, P.G., Kuhn, E.M. and Therman, E. (1987). What causes the abnormal phenotype in a 49,XXXXY male? Hum. Genet.76: 1-4.
- Swillen, A., Devriendt, K., Legius, E., Eyskens, B., Dumoulin, M., Gewillig, M. and Fryns, J.P. (1997). Intelligence and psychosocial adjustment in velocardiofacial syndrome: a study of 37 children and adolescents with VCFS. J. Med. Genet.34: 453-458.
- Willard, H. (1996). X-chromosome inactivation and X-linked mental retardation. Am. J. Med. Genet.64: 21-26.
- Yokoyama, Y., Narahara, K., Teraoka, M., Koyama, K., Seino, Y., Yagi, S., Konishi, T. and Miyawaki, T. (1997). Cryptic pericentric inversion of chromosome 17 detected by fluorescence in situ hybridization study in familial Miller-Dieker Syndrome. Am. J. Med. Genet. 71: 236-237.
Publication Dates
-
Publication in this collection
06 Jan 1999 -
Date of issue
June 1998
History
-
Received
11 May 1998