Abstract
Several new microdeletion and microduplication syndromes are emerging as disorders that have been proven to cause multisystem pathologies frequently associated with intellectual disability (ID), multiple congenital anomalies (MCA), autistic spectrum disorders (ASD) and other phenotypic findings. In this paper, we review the "new" and emergent microdeletion and microduplication syndromes that have been described and recognized in recent years with the aim of summarizing their main characteristics and chromosomal regions involved. We decided to group them by genomic region and within these groupings have classified them into those that include ID, MCA, ASD or other findings. This review does not intend to be exhaustive but is rather a quick guide to help pediatricians, clinical geneticists, cytogeneticists and/or molecular geneticists.
microdeletion; microduplication; chromosome rearrangement; novel deletions; novel duplications
REVIEW ARTICLE
New microdeletion and microduplication syndromes: a comprehensive review
Julián NevadoI,II,* * These authors contributed equally to this work. ; Rafaella MergenerIII,* * These authors contributed equally to this work. ; María Palomares-BraloI,II; Karen Regina SouzaIII; Elena VallespínI,II; Rocío MenaI,II; Víctor Martínez-GlezI,II; María Ángeles MoriI,II; Fernando SantosI,IV; Sixto García-MiñaurI,IV; Fé García-SantiagoI,V; Elena MansillaI,V; Luis FernándezI,VI; María Luisa de TorresI,V; Mariluce RiegelIII,VII,$ $ These authors contributed equally to this work. ; Pablo LapunzinaI,IV,VIII,$ $ These authors contributed equally to this work.
ICentro de Investigación Biomédica en Red de Enfermedades Raras, Instituto de Salud Carlos III, Madrid, Spain
IISection of Functional and Structural Genomics, Instituto de Genética Médica y Molecular, Hospital Universitario la Paz, Madrid, Spain
IIIPrograma de Pós-graduação em Genética e Biologia Molecular, Universidade Federal do Rio Grande do Sul, Porto Alegre,RS, Brazil
IVSection of Clinical Genetics, Instituto de Genética Médica y Molecular, Hospital Universitario la Paz, Madrid, Spain
VSection of Cytogenetics, Instituto de Genética Médica y Molecular, Hospital Universitario la Paz, Madrid, Spain
VISection of Preanalytics, Instituto de Genética Médica y Molecular, Hospital Universitario la Paz, Madrid, Spain
VIIServiço de Genética Médica, Hospital de Clínicas de Porto Alegre, Porto Alegre ,RS, Brazil
VIIISection of Molecular Endocrinology, Overgrowth Disordes Laboratory, Instituto de Genética Médica y Molecular, Hospital Universitario la Paz, Madrid, Spain
Send correspondence to Send correspondence to: Pablo Lapunzina Instituto de Genética Médica y Molecular, Hospital Universitario la Paz Paseo de la Castellana 261-28046, Madrid, Spain E-mail: plapunzina.hulp@salud.madrid.org.
ABSTRACT
Several new microdeletion and microduplication syndromes are emerging as disorders that have been proven to cause multisystem pathologies frequently associated with intellectual disability (ID), multiple congenital anomalies (MCA), autistic spectrum disorders (ASD) and other phenotypic findings. In this paper, we review the "new" and emergent microdeletion and microduplication syndromes that have been described and recognized in recent years with the aim of summarizing their main characteristics and chromosomal regions involved. We decided to group them by genomic region and within these groupings have classified them into those that include ID, MCA, ASD or other findings. This review does not intend to be exhaustive but is rather a quick guide to help pediatricians, clinical geneticists, cytogeneticists and/or molecular geneticists.
Keywords: microdeletion, microduplication, chromosome rearrangement, novel deletions, novel duplications.
Introduction
Alteration of gene dosage due to gains or deletions of large genomic regions causes many genetic disorders that are frequently associated with intellectual disability (ID), multiple congenital anomalies (MCA), autistic spectrum disorders (ASD) and other phenotypic findings (Lupski and Stankiewicz, 2005) The advances in the use of microarrays for diagnosis and research in genomic disorders has permitted the discovery of infrequent genomic rearrangements in a variety of diseases and the report of several microdeletion and microduplication syndromes (Deak et al., 2011; Rafati et al., 2012; Vissers and Stankiewicz , 2012;.Weise et al., 2012).
The identification of novel syndromes is based on consistent, clinically recognizable features associated with a common chromosomal region; however, for some copy number variations (CNVs), variability in expression and penetrance of clinical manifestations have complicated the establishment of their clinical significance. Currently, the delineation of novel syndromes may start with the identification of overlapping genotypes, that is, a 'genotype-first' approach, in which patients are characterized by a similar genomic aberration before a common clinical presentation is delineated. This is referred to as "reverse genetics". This approach has proven to be successful considering the growing list of microdeletion/microduplication syndromes that have been described in the past five years. Furthermore, the collection of clinical and genetic information in databases such as DECIPHER, ISCA, ECARUCA and other free databases has been crucial for discriminating between patients with rare aberrations and those with new microdeletion/duplication syndromes.
In this paper, we systematically review the novel microdeletion and microduplication syndromes described in the past five years. We grouped these disorders by chromosome, with the intention of serving as a quick guide for clinicians and researchers.
Chromosome 1
1p34.2-p34.3 Deletion. This deletion is characterized by microcephaly, ID and ASD. The deletion spans approximately 3.3 Mb and involves approximately 43 genes, including RIMS3, which is the main candidate gene for this phenotype.
1p31.3-p32.2 Deletion. There are approximately 7 cases described to date. In five of these cases with an NFIA deletion, the patients show central nervous system (CNS) malformations (hypoplasia of the corpus callosum, macrocephaly, ventriculomegaly) and urinary tract defects.
1q21.1 Deletion/Duplication. Patients have a phenotype similar to 22q11.2 deletion syndrome (velocardiofacial syndrome-like phenotypic findings). They may have congenital heart disease, schizophrenia and ID. The microduplication of approximately 212 kb could be responsible for congenital heart disease in at least 2 patients. The phenotype of dup1q21.1 is variable due to incomplete penetrance and variable expression levels. Therefore, this microduplication is also observed in asymptomatic individuals. Patients with deletions (1.4-1.65 Mb) may also have microcephaly, epilepsy, ataxic gait, severe dysmorphic features of the face and ID. This deleted region comprises approximately 30 coding genes, including the cluster of genes encoding the ephrins (EFNA1, EFNA3 and EFNA4), which are tyrosine kinase receptors.
1q24-q25 Deletion. This deletion is characterized by growth retardation, microcephaly, small hands and feet (with brachydactyly), dysmorphic facies, small ears, micrognathia, short nose with bulbous tip and severe ID. The deleted region is approximately 1.9 Mb (chr1: 170135865-172099327 coordinates hg18) and contains 13 genes including DNM3 and CENPL, which encodes a protein essential for centromeric function, mitotic progression and synaptic reaction.
1q32.2-q32.3 Deletion. Patients exhibit dysmorphic features and facial clefts due to deletion of the IRF6 gene, which is responsible for the Van der Woude syndrome (VWS). The deletion is approximately 2.98 Mb and includes 25 genes.
1q41-q42.12 Deletion. This deletion is characterized by moderate to severe ID, seizures, Pelger-Huet anomaly (leukocyte alteration), cleft lip and palate and agenesis of the corpus callosum. Patients may also have hypoglycemia, 13 pairs of ribs and a micropenis. DISP1, in the sonic hedgehog pathway, has been proposed as the gene responsible for the alterations of the midline observed in this deletion. The deletions have a size of 777 kb to 6.87 Mb. It has been proposed that this would represent a locus for Fryns syndrome, a Fryns syndrome phenocopy, or congenital diaphragmatic hernia (CDH). It has also been observed in patients with the isolated 1q42 deletion (together with agenesis of the corpus callosum) or within a contiguous deletion, 1q41q42 syndrome (Filges et al., 2010).
1q43-q44 Deletion. Microcephaly, abnormalities of the corpus callosum, seizures, ID and speech disorder are observed in patients with this deletion. Deletion of the AKT3 gene appears to be correlated with microcephaly and alteration of the other 3 genes (FAM36A, C1ORF199 and HNRNPU) correlates with the epileptic phenotype.
Chromosome 2
2p14-p15 Deletion. This deletion is associated with ID, speech disorder, mild dysmorphic features, hearing loss and relative microcephaly. The deletion is approximately 2.23-2.84 Mb, with a minimal overlapping region of 10 genes.
2p15-q16.1 Deletion. This deletion is associated with ID, ASD and dysmorphic features. Eight patients described by three different groups have this deletion. The OTX1 and XPO1 genes have been associated with ASD. It has also been observed in patients with prenatal and postnatal growth retardation, ptosis of both eyelids and microcephaly. The deletions range from 2.6 Mb to 3.2 Mb.
2q13 Deletion. Patients with this deletion have CNS disorders and present with cortical disruption and Joubert syndrome. These phenotypes are associated with the deletion of the NPHP1 gene.
2q23.1 Deletion/Duplication. Seizures, speech disturbances, ataxia, short stature, ID and dysmorphic features (brachycephaly, proximal implementation of the hair, short nose, hypertelorism, everted lower lip, thick tongue, brachytelephalangy, clinodactyly and hypertrichosis) are observed. This deletion includes the MBD5 gene, which is implicated in the pathophysiology of seizures. Stereotyped behaviors are also present and these are similar to Rett syndrome or Angelman syndrome. Microduplications in this region present with ID, hypotonia, ASD and include the following genes: MBD5, ACVR2A, ORC4L, EPC2, KIF5C, MIR1978, LYPD6B and LYPD6. (Jaillard et al., ,2009).
2q31.1-q31.2. Deletion. This deletion is characterized by monodctylous hands, ectrodactyly, brachydactyly and clinodactyly with or without duplication of both halluces. This deletion is of the HOXD cluster. Mutations in HOXD13 and HOXD10 are associated with malformations of the limbs. Some patients may have ID, microcephaly and growth retardation.
2q32-33.1 Deletion. Patients with this deletion have ID, learning disabilities, growth retardation, thin and sparse hair, feeding difficulties, cleft lip/palate and multiple dysmorphic features. SATB2 haploinsufficiency has been suggested to be responsible for most of these findings. The deletions are between 35 kb to 10.4 Mb (Rosenfeld et al., 2009)].
2q37 Deletion. This deletion shows the Albright phenotype (hereditary osteodystrophy; brachydactyly, ID and short stature).
Chromosome 3
3p21.31 Deletion. Patients present with cortical blindness, CNS abnormalities, cleft lip and ID. The deletions are approximately 3.1 Mb, including approximately 80 genes.
3p25 interstitial Deletion. Patients present with low birth weight, mental retardation, telecanthus, ptosis and micrognathia and congenital heart disease, typically atrioventricular septal defect. SRGAP3 is the major determinant of ID.
3p11.2-p12.1 Deletion. This deletion includes POU1F1, CHMP2B and VGLL3. Patients have abnormalities in pituitary hormones similar to the hypothalamic Laron syndrome but are unresponsive to GH treatment.
3q13.31 Deletion. ID, postnatal overgrowth, hypoplastic genitalia (in men) and recognizable facial features (short philtrum and protruding lips) are observed in these patients. The deletion is 580 kb and includes DRD3 and ZBTB20 as candidate genes.
3q22.1-q25.2 Deletion. Patients with this deletion have multiple congenital anomalies and peculiar facial appearance. In particular, the phenotypes result from the variable combination of three recognizable patterns: Dandy-Walker malformation, BPES syndrome and Wisconsin syndrome.
3q27.3 Deletion. Patients shared a recognizable facial dysmorphism and Marfanoid habitus associated with psychosis and mild to severe ID. Most of these patients have severely impaired adaptive skills.
3q29 Deletion/Duplication. Facial dysmorphism, ASD, psychiatric disorders (bipolar disease), ID and MCA (cleft palate, congenital heart disease) are all associated with deletions in this region. There are reports of patients with deletions and parents with mosaic deletions. The microduplication is characterized by mild ID, microcephaly, dysmorphic features and musculoskeletal abnormalities. The minimum size of the rearrangement is 1.6 Mb (Figure 1A).
Chromosome 4
4q21 Deletion. Growth retardation, ID and absence of language are found in patients with this deletion. The smallest region of overlap in deletions is 1.37 Mb and contains five genes: PRKG2, RASGEF1B, HNRNPD, HNRPDL and ENOPH1. It has been suggested that PRKG2 and RASGEF1B are the genes responsible for this clinical phenotype.
4q21.3 Deletion. Patients present ID, dysmorphic facial features, hypotonia and short stature.
4q34.1-q35.2 Deletion. The phenotype is somewhat similar to the 22q11.2 deletion syndrome.
Chromosome 5
5q14.3-q15 Deletion. ID (late Honest), epilepsy, hypotonia and dimples in the jugular region are found in patients with this deletion. Patients present with atypical clinical Rett syndrome features. The gene involved is MEF2C.
5q35.2-q35.3 Deletion/Duplication. The deletion is
1.63 Mb and sometimes includes NSD1, the gene responsible for Sotos syndrome. Patients also have the Sotos phenotype, cleft palate, language delay, ID and macrocephaly. Microduplication of the Sotos syndrome region, which contains NSD1, has been associated with microcephaly, short stature and development delay.
Chromosome 6
6p25 Deletion. Patients present with white matter abnormalities, hypotonia, ID, a dysmorphic face, hypoacusis, short stature, Axenfeld-Rieger anomaly and a bicuspid aortic valve.
6q13-q14 Deletion. ID and connective tissue abnormalities are found in patients with this deletion. The deletion is 3.7 Mb and affects 16 genes, including COL12A1,a good candidate for the anomaly in the connective tissue.
6q14.1-q15 Deletion. Obesity, ID and atypical facial phenotypic traits are observed among these patients. This deletion syndrome partly phenocopies patients with Prader-Willi syndrome. The haploinsufficiency of SIM1 is suggested to be responsible for the phenotype.
6q16.1 Deletion. Patients present with ID and characteristic facies. The deletion includes the haploinsufficiency of one gene (ephrin receptor 7; EPHA7) that has implications in cortical development.
6q25 Deletion. Patients usually show ID. Olfactory bulb aplasia and anosmia may also be observed. The syndrome is due to the haploinsufficiency of ARID1B, a member of the SWI/SNF chromatin-remodeling complex. Some patients with ID have point mutations in this gene.
6q25.2-q25.3 Deletion. Microcephaly, ID, hypoacusis and dysmorphic features are observed in patients with this deletion. The smallest region of overlap of the deletion in 4 patients is 3.52 Mb in size.
Chromosome 7
7p14.1 Deletion. This deletion is characterized by polysyndactyly, hypertelorism and microcephaly, having a phenotype similar to Greig syndrome.
7p22.1 Duplication. Speech delay and recognizable facial features are observed in these patients. The duplication is approximately 1.7 Mb. Macrocephaly, ocular hypertelorism and low-set ears can also occur. Fifteen genes are involved in the duplicated segment.
7q11.23 Duplication. It is the reciprocal duplication of the deletion observed in Williams Syndrome. Patients present with speech delay, epilepsy, ID, straight and thick eyebrows and ASD.
7q11.23 Deletion. This is the distal deletion of the Williams-Beuren region. The deletions are recurrent, 1.2 Mb in size and include the Huntingtin-interacting protein 1 (HIP1) and tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein gamma (YWHAG) genes. The deletion of HIP1 seems to be sufficient to cause ID.
7q21.3 Deletion. Myoclonus, dystonia, ID and psychosis have been observed in these patients. Two regions of 455 and 496 kb are critical for ID, which is where the gene LOC253012 (HEPACAM2) is located.
7q22.2-q22.3 Deletion. Overgrowth, delayed bone age, epilepsy, ID, unusual face, hypoplasia of the corpus callosum and cerebellar hypoplasia are observed in patients with this deletion. The deletion spans 3.2 Mb and includes four of 15 genes involved in cell cycle (SRPK2, MLL5, RINT1 and LHFPL3).
7q33-q35 Deletion. Patients with this deletion present with speech delay and ID. The deletion includes CNTNAP2, a gene that has previously been found in children with speech disorders.
Chromosome 8
8p21 Deletion. ID and behavior disorder, with some features of autistic spectrum disorder occur in patients with this deletion.
8p23.1 Duplication. This duplication leads to a variable phenotype that may include one or more of the following: congenital heart disease (CHD), ID and mild dysmorphism with prominent forehead and arched eyebrows. The critical region is a duplication of 3.68 Mb that contains 31 genes and microRNAs, of which only GATA4, TNKS, SOX7 and XKR6 are likely to be dosage-sensitive genes. Of the microRNAs, MIR124-1 and MIR598 have been implicated in neurocognitive phenotypes. A combination of the duplication of GATA4, SOX7 and related genes may account for the variable penetrance of CHD.
8q12 Duplication. This duplication includes CHD7. Patients present with hypotonia, ID, failure to thrive, Duane anomaly, Mondini malformation, hearing loss, malformations of the ear canal and atrial septal defects.
8q21.11 Deletion. ID, round face with full cheeks, eyelid ptosis, ocular malformations, hypoplastic nose, abnormal philtrum and vermillion and minimal hand anomalies (camptodactyly, syndactyly) are all observed in these patients. The smallest region of overlap is 539.7 kb, which includes three genes, including Zinc Finger Homeobox 4 (ZFHX4)(Figure 1B).
8q22.1 Deletion. The phenotype of this deletion is similar to that of the Nablus mask syndrome, symptoms include ID, speech disorder and typical dysmorphic features. The deletion is approximately 1.6 Mb.
8q22.2 Deletion. Patients with this deletion have characteristic facial features, ID, absent speech, seizures, growth retardation and diaphragmatic hernia in some cases. The smallest region of deletion is 3.87 Mb (100.69 to 104.56 Mb, hg18), comprising at least 25 genes.
Chromosome 9
9p13.3-p13.1 interstitial Deletion. Patients with a 9p13 deletion have mild to moderate ID, social and interactive personality and behavioral problems, such as attention deficit-hyperactivity disorder. Short stature, prominent antihelices, hypoplastic nails and precocious/early puberty are also present (Figure 1C).
9q22.3 Deletion. ID, dysarthria, metopic craniosynostosis, hydrocephalus, macrosomia and seizures are associated with this deletion. Patients with a 9q22.3 microdeletion have the clinical findings of the Gorlin syndrome. The 9q22.3 microdeletions (352 kb to 20.5 Mb in size) include PTCH1, the gene that is mutated in Gorlin syndrome (nevoid basal cell carcinoma syndrome).
9q31.1-q31.3 Deletion Patients have a distinct clinical phenotype characterized by mild ID, short stature with high body mass index, thick hair, arched eyebrows, flat profile with broad chin, mild prognathism, broad and slightly overhanging tip of the nose and short neck with cervical gibbus.
9q34.3 Deletion (Kleefstra syndrome). Patients have ID, behavior anomalies, hypotonia, epilepsy, congenital heart defect and renal anomalies. The highly recognizable facial features are hypertelorism, midface hypoplasia, prognathism, prominent eyebrows, cupid bow or tented upper lip and everted lower lip. The syndrome is either caused by a submicroscopic deletion in the chromosomal region 9q34.3 or an intragenic mutation of the EHMT1 gene causing haploinsufficiency of EHMT1. There are some communications of mosaicism in parents of affected patients (Figure 1D).
Chromosome 10
10q22-q23 Deletion. The overlap of neuropathological phenotypes among patients described suggests that this region harbors genes important for function and neurodevelopment (NRG3, GRD1D1, BMPR1A, SNCG). Indeed, several genes in this region are candidates for neuropsychiatric disorders. NRG3 and GRD1D1 have been described as candidate genes associated with schizophrenia.
Chromosome 11
11q13.1 Deletion. Speech delay, autistic spectrum disorder, dysmorphic features, such as wide halluces and firsts digits and pancreatic gastrinoma. The deletion is 0.57 Mb.
Chromosome 12
12q13.11 Deletion. Severe ID, cleft palate and severe myopia. The deleted region contains 16 genes. It is hypothesized that haploinsufficiency of AMIGO2 is responsible for the ID and haploinsufficiency of COL2A1 in cleft palate and myopia.
12q13 Duplication. This syndrome may represent a phenocopy of the Wolf Hirschhorn syndrome (Bertioli et al., 2013)
12q14 Deletion. Microcephaly, short stature with similar clinical findings to the Russell Silver syndrome. Patients may have osteopoikilosis, low weight, failure to thrive and learning problems. The HMGA2 gene is involved in the growth deficiency.
12q24.31 Deletion. Hypoglycemia, macroglossia and overgrowth (similar to the Beckwith-Wiedemann syndrome at birth). The deleted region contains the gene HNF1 homeobox A (HNF1A) and others.
Chromosome 14
14q12-q22.1 Deletion. Patients present ID, failure to thrive, microcephaly and recognizable facial features (hypertelorism, epicanthic folds, peculiar eyebrows, depressed nose, receding forehead, CNS disorders, seizures, apnea, myoclonus and infection proneness).
14q32.2 Deletion. ID and many phenotypic abnormalities in two unrelated patients with identical deletions. The deletions are mediated by repetitions (TGG) (n)
14q32.33 Deletion. ID and minimal dysmorphic features. The deletion affects a small fragment of 0.3 Mb with 6 genes, including NUDT14, BRF1, BTBD6, PACS2, MTA1 and TEX22. A 250-kb region critical for certain features of terminal 14q deletion syndrome has been proposed. Amongst them three potential candidate genes for intellectual disability: CRIP2, MTA1 and TMEM121 (Engels et al., 2012).
Chromosome 15
15q11.2 Deletion. ID, speech delay, behavioral problems, seizures and ASD. The deletion is between the BP1 and BP2 regions of the proximal portion of chromosome 15 that contains four genes (TUBGCP5, NIPA1, NIPA2 and CYFIP1) not subjected to imprinting.
15q11-q13 Duplication. ASD, mild facial dysmorphism, sleep problems and unusual electroencephalogram findings (Urraca et al., 2013).
15q13.2-q13.3 Deletion. Phenotypic findings similar to the Angelman syndrome, with ASD, epilepsy and behavioral problems.
15q13.3 Deletion. Epilepsy, ID, psychiatric disorders (bipolar disorder), severe hypotonia and EEG abnormalities. Locus with incomplete penetrance for autism; may show retinal dysfunction and encephalopathy. One gene appears to be involved (CHRNA7) (Shinawi et al., 2009).
15q14 Deletion. Dandy-Walker malformation, ID, macrocephaly, myopia and brachytelephalangy.
15q21.1-q21.2 Deletion. Clinical features similar to Marfan syndrome and with ID. The deletions involve the FBN1 gene.
15q24 Deletion/Duplication. Growth delay, ID, facial features (long face, anterior hairline, epicanthic folds, hypertelorism, long philtrum and thick lower lip). Other findings include ASD, hypotonia, behavioral problems, hearing loss, hernias and GH deficiency. Most deletions have breakpoints in five LCRs (LCR15q24A, -B, -C, -D and -E) and the minimum region of overlap is 1.2 Mb between LCR15q24B and LCR15q24C. Candidate genes within this deletion are CYP11A1, SEMA7A, CPLX3, ARID3B, Stra6, Sin3A and CSK. The duplication cases described share similar clinical features with the 15q24 deletion.
15q24.1 Deletion. Multiple cysts of the corpus callosum, ID, micropenis and strabismus. The deletion is approximately 3.1 Mb. The sizes of the deleted regions range from 1.7 Mb to 6.1 Mb. Most of the reported cases are male. Male genital abnormalities are frequently observed in 15q24 microdeletion patients. One candidate gene is CYP11A1, which is highly expressed in the adrenal gland.
15q24.3-q25.2 Deletion. Cleft palate with or without cleft lip and hypotonia.
15q26 Deletion. Patients have mainly short stature due to haploinsufficiency of the IGF1R gene.
15q26.1 Deletion. Intractable epilepsy, ID and short stature.
Chromosome 16
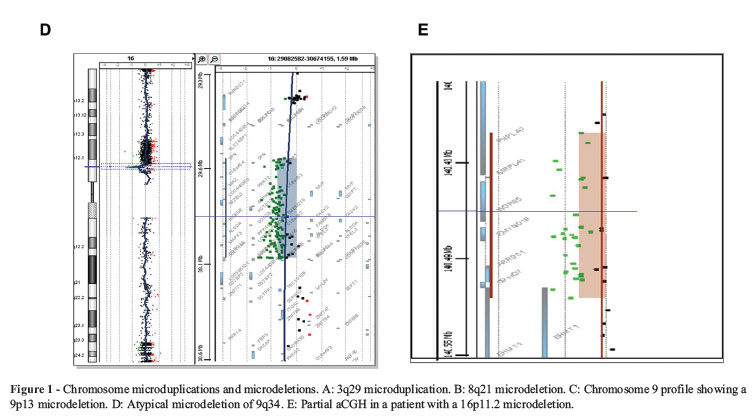
16p11.2 Deletion/Duplication. The deletion is characterized by ID, ASD, epilepsy and other less common findings, such as obesity, microphthalmia, coloboma of the optic nerve, kidney and urinary tract abnormalities, Hirschsprung disease, endocardial fibroelastosis and hemivertebrae. Duplication is associated with autism, ID, CNS disorders and schizophrenia (Rosenfeld et al., 2010). A 600-kb 16p11.2 deletion containing 29 genes has been associated with several neurocognitive disorders, including autism, diabetes-independent obesity and microcephaly, whereas duplication of the same region is associated with autism, schizophrenia, anorexia and microcephaly. The 16p11.2 deletion is associated with increased head size, whereas 16p11.2 duplication is associated with decreased head size (Golzio et al., 2012) (Figure 1E).
16p11.2-p12.2 Deletion/Duplication. Minimal facial abnormalities, speech disorder, frequent ear infections and ID. It should be distinguished from the proximal deletion (see immediately above). Patients with duplications have severe ID, ASD and dysmorphic features (Figure 2A and B).
16p12.1 Deletion. ID and abnormal behavioral phenotype with behavioral disorders. It is a 520-kb deletion.
16q12-q13 Deletion. The phenotypic spectrum of microdeletions in 16q12-q13 region is broad with variable degrees of ID, craniofacial dysmorphic features, congenital brain abnormalities and limb and congenital heart disease.
16q22.1 Deletion/Duplication. Deletion presents ID and lobular breast cancer. The deletion is 0.24 MB and affects 3 genes (ZFP90, CDH3 and CDH1). ZFP90 is expressed in the brain and is responsible for ID, while CDH1 may be responsible for cancer. Duplication is characterized by epilepsy and learning disabilities.
16q24.1 Deletion. Typically present persistent pulmonary hypertension in the newborn and sometimes atrioventricular canal, ureteral stenosis and annular pancreas. FOXF1 alterations would be responsible for alveolar capillary dysplasia and other alveolar malformations.
16q24.3 Deletion. ID, ASD, short stature and minimal facial anomalies. Deletions involve the ANKRD11 gene and cause KBG-like syndrome.
Chromosome 17
17p13.1 Deletion. ID, hypotonia and anomalies of the hands and feet but not cancer. Microdeletions affect the TP53 gene.
17p13.3 Deletion/Duplication. Short stature, ID and abnormal facial features. The microduplication includes autism and affects a region of 72 kb that includes a single gene (YWHAE). The microdeletion sometimes includes CRK, sometimes YWHAE and/or TUSC5 (Figure 2C and 2D).
17q11.2 Deletion/Duplication. The deletion and duplication affects the NF1 region. ID, facial dysmorphic features and seizures.
17q12 Deletion/Duplication. The deletion presents congenital diaphragmatic herniae and pulmonary and renal cysts. A case of Mayer-Rokitansky-Kuster-Hauser syndrome has been described with this deletion. The deletion is
1.4 Mb and affects 17 genes, including AATF, ACACA, DDX52, DUSP14, GGNBP2, HNF-1B, Lhx1, PIGW, SYNRG, TADA2A and ZNHIT3. Duplication is characterized by ASD (Figure 2E).
17q21.31 Deletion/Duplication. Deletions are associated with macrocephaly, ID, epilepsy, congenital anomalies and dysmorphic facial alterations of the pituitary. The skin lesions are characterized by nevi, abnormal skin pigmentation similar to cardiofaciocutaneous syndrome. Other findings include dilation of the aortic root, joint subluxation, hearing loss, recurrent otitis media and persistent digital pads. At least 6 genes are affected, including MAPT and STH.
17q22-q23.2 Deletion. Microcephaly, thyroid duct cyst, sensorineural hearing loss and pulmonary hypertension. Includes the loss of TBX2 and TBX4 but not NOG.
17q23.1-q23.2 Deletion/Duplication. Duplication has been associated with pes cavus familiar. Duplication affects PITX1 and TBX4. The deletion has congenital heart disease and limb abnormalities.
17q23.2 Deletion. Bilateral sensorineural hearing loss in two isolated patients.
17q24.2-q24.3 Deletion/Duplication. Duplication presents generalized hypertrichosis with gingival hyperplasia and deletions in general have less gingival hyperplasia.
Chromosome 18
18q12.3 Deletion. The deletion is 372 kb in size with haploinsufficiency of SETBP1. The clinical findings include ID and speech disorder. Missense heterozygous mutations in this gene cause Schinzel-Giedion syndrome (SGS). However, the phenotype of individual with partial chromosome 18q deletions does not resemble SGS.
Chromosome 19
19p13.11 Deletion. One patient presented with pontocerebellar hypoplasia and ID and haploinsufficiency of the helicase DDX39. Another patient with a deletion in this region showed a deletion of 1.1 Mb involving EPS15L1. The patient showed short stature, ID, severe hypotonia, ataxia, premature pubarche and dysmorphic features.
19p13.12 Deletion. Defects of the branchial arches (preauricular tags, ear canal stenosis), mild hearing loss and mild ID are due to the deletion of a region of 0.8 Mb of genomic DNA. The deletion extends 15300338-16064271 (hg18, NCBI build 36.1). One patient presented with ID, obesity and hypertrichosis.
19p13.13 Deletion/Duplication. The deletion is characterized by macrocephaly, overgrowth and ophthalmologic and gastrointestinal disturbances. Duplication has short stature and microcephaly. The smallest region of overlap is 311-340 Kb and has 16 genes including MAST1, NFIX and CALR.
19p13.2 Deletion. Patients present with ID, mild facial features, febrile seizures. The deletion is 834.2 kb in size and includes 32 genes.
19p13.3 Terminal Duplication. Patients' phenotypes include severe psychomotor DD, skeletal malformations and a distinctive facial appearance.
19q13.2 Deletion. Patients present branchial arch defects. The critical region is approximately 0.8 Mb
19q12-q13.2 Duplication. It is an obesity-related syndrome, with ID and minor facial findings.
19q13.11 Deletion. Patients present a Diamond-Blackfan syndrome, pre and postnatal growth deficiency, tall stature, microcephaly, hypospadias, signs of ectodermal dysplasia and aplasia cutis vertex. The critical region is defined to 750 kb and is due to haploinsufficiency of RPS19 (Malan et al., 2009).
Chromosome 20
20p12.3 Deletion. Cleft palate / cleft lip, Pierre-Robin sequence. The deletion involves the BMP2 gene and has been implicated in Wolff-Parkinson-White (WPW) syndrome with neurocognitive deficits and with Alagille syndrome when the deletion includes the neighboring JAG1 gene in addition to BMP2.
20p13 Deletion. Dysmorphic features, ID, epilepsy and brachydactyly. SOX12 and NRSN2 are the candidate genes that may be involved in the developmental defects.
20q13.33 Deletion. Severe limb malformations, skeletal abnormalities, ID, speech delay, seizures and other minimal dysmorphic features. The ARFGAP1, CHRNA4 and KCNQ2 genes have been associated with neurological deficits.
Chromosome 21
21q22 Deletion. Two patients with similar phenotype and overlapping deletions of the 21q22 region were observed. They had behavioral problems, no speech, microcephaly, feeding problems, regurgitation, obesity dysplastic ears and pointed chin. They also present with cerebral atrophy, thinned corpus callosum, epilepsy and ventricular septal defect. Another patient had microcephaly, ID, hypospadias and corneal opacity. Patients with chromosomal deletions of 21q show a variation in size and usually include the 21q22.12 region. The deleted region in most of these patients included, among other deleted genes, the RUNX1 gene (21q22.12), which is related with thrombocytopenia and platelet function and a predisposition to develop myeloid leukemia.
Chromosome X
Xq11.11 Deletion. Patients with this deletion have ID, epilepsy, macrosomia, macrocephaly, tall stature and dysmorphic features. The 1.3 Mb deletion includes ARHGEF9, which is proposed to have a role in the cognitive development.
Conclusions
The extensive use of high-resolution microarrays and the "genotype-first" experimental approach taken by genetic laboratories have allowed for the recognition and description of an important and growing number of new microdeletion and microduplication syndromes over the last three to five years (Figure 3). Interestingly, most of these syndromes have phenotypic features that are similar to and overlapping with other previously described genetic syndromes. This review may be considered a quick guide to help pediatricians, clinical geneticists, cytogeneticists and/or molecular geneticists and emphasizes the necessity of a strong collaboration between clinical (pediatricians, geneticists) and molecular geneticists to assure new phenotype recognitions for these genomic aberrations.
License information: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Bertoli M, Alesi V, Gullotta F, Zampatti S, Abate MR, Palmieri C, Novelli A, Frontali M and Nardone AM (2013) Another patient with 12q13 microduplication. Am J Med Genet A 161A:2004-2008.
- Deak KL, Horn SR and Rehder CW (2011) The evolving picture of microdeletion/microduplication syndromes in the age of microarray analysis: Variable expressivity and genomic complexity. Clin Lab Med 31:543-564.
- Engels H, Schüler HM, Zink AM, Wohlleber E, Brockschmidt A, Hoischen A, Drechsler M, Lee JA, Ludwig KU, Kubisch C, et al. (2012) A phenotype map for 14q32.3 terminal deletions. Am J Med Genet A.158A:695-706.
- Filges I, Röthlisberger B, Boesch N, Weber P, Wenzel F, Huber AR, Heinimann K and Miny P (2010) Interstitial deletion 1q42 in a patient with agenesis of corpus callosum: Phenotype-genotype comparison to the 1q41q42 microdeletion suggests a contiguous 1q4 syndrome. Am J Med Genet A 152A:987-993.
- Jaillard S, Dubourg C, Gérard-Blanluet M, Delahaye A, Pasquier L, Dupont C, Henry C, Tabet AC, Lucas J, Aboura A, et al. (2009) 2q23.1 microdeletion identified by array comparative genomic hybridisation: An emerging phenotype with Angelman-like features? J Med Genet 46:847-855.
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. (2012) KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature.485:363-367.
- Lupski, J and Stankiewicz, P (2005) Genomic disorders: Molecular mechanism for rearrangements and conveyed phenotypes. PLoS Genetics 1:e-49.
- Malan V, Raoul O, Firth HV, Royer G, Turleau C, Bernheim A, Willatt L, Munnich A, Vekemans M, Lyonnet S, et al. (2009) 19q13.11 deletion syndrome: A novel clinically recognisable genetic condition identified by array comparative genomic hybridisation. J Med Genet 46:635-640.
- Rafati M, Seyyedaboutorabi E, Ghadirzadeh MR, Heshmati Y, Adibi H, Keihanidoust Z, Eshraghian MR, Javadi GR, Dastan J, Mosavi-Jarrahi A, et al. (2012) "Familial" vs. "Sporadic" intellectual disability: Contribution of common microdeletion and microduplication syndromes. Mol Cytogenet 5:9.
- Rosenfeld JA, Ballif BC, Lucas A, Spence EJ, Powell C, Aylsworth AS, Torchia BA and Shaffer LG (2009) Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS One 4:e6568.
- Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE, Shaffer LG and Ballif BC (2010) Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications J Neurodev Disord 2:26-38.
- Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, Lanpher B, Nagl S, Herding HS, Nevinny-Stickel C, et al. (2009) A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet 41:1269-1271.
- Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, Schanen NC, Esmer C, Lamport D and Reiter LT (2013) The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res 6:268-279.
- Vissers LE and Stankiewicz P (2012) Microdeletion and microduplication syndromes. Methods Mol Biol 838:29-75.
- Weise A, Mrasek K, Klein E, Mulatinho MV, Llerena JC Jr, Hardekopf D, Pekova S, Bhatt S, Kosyakova N and Liehr T (2012) Microdeletion and Microduplication Syndromes. J Histochem Cytochem 60:346-358.
Publication Dates
-
Publication in this collection
21 May 2014 -
Date of issue
2014