Abstracts
The absorption spectra of DPH at fixed concentration do not change with water content in organic solvents. It exhibits monomer bands, such as those obtained in ethanol. The absorption did not change for solutions up to 54 and 46% of water in ethanol and DMSO, respectively, for [DPH] = 5.0 × 10-6 mol L-1 at 30 °C. However, at the same experimental conditions, a gradual sharp decay of the DPH fluorescence is observed. It is proposed that water molecules below these water concentration limits act as quenchers of the excited states of DPH. Stern-Volmer quenching constants by intensities measurements are 7.4 × 10-2 (water/ethanol) and 2.6 × 10-2 L mol-1 (water/DMSO). DPH lifetime measurements in the absence and presence of water resulted in 7.1 × 10-2 L mol-1 in water/ethanol, which pointed out that the process is a dynamic quenching by water molecules. For experiments using DPH as probe, this process can affect data, leading to misunderstanding interpretation.
diphenylhexatriene; DPH; DPH fluorescence; dynamic quenching; lifetime
O espectro de absorção de DPH, em concentração fixa, não varia com o teor de água em solvente orgânico. Tem-se a banda de monômeros igual àquela em etanol puro. A absorção não muda até o limite de 54 e 46% de água em etanol e DMSO, respectivamente, para [DPH] = 5,0 × 10-6 mol L-1 a 30 °C. Entretanto, em misturas com água muito abaixo desses conteúdos críticos, observou-se um decaimento intenso de fluorescência enquanto a absorção manteve-se constante. Propõe-se que moléculas de água atuam como supressores dos estados excitados e a constante de supressão de Stern-Volmer através de intensidade relativas, resultou em 7,4 × 10-2 (água/etanol) e 2,6 × 10-2 L mol-1 (água/DMSO). Os tempos de vida do DPH na ausência e presença do supressor forneceram constantes de 7,1 × 10-2 L mol-1 em água/etanol, indicando supressão dinâmica. Em investigações de ambientes com esta sonda, este processo deve ser considerado tendo em vista o risco de erros de interpretação.
ARTICLE
Unusual 1,6-diphenyl-1,3,5-hexatriene (DPH) spectrophotometric behavior in water/ethanol and water/DMSO mixtures
Augusto C. GracettoI; Vagner R. BatistelaI; Wilker CaetanoI; Hueder P. M. de OliveiraII; Willy Glen SantosIII; Carla C. S. CavalheiroIII; Noboru HiokaI, * * e-mail: nhioka@uem.br
IDepartamento de Química, Universidade Estadual de Maringá, Av. Colombo 5.790, 87020-900 Maringá-PR, Brazil
IIUniversidade Camilo Castelo Branco, São José dos Campos, 12247-004 São Paulo-SP, Brazil
IIIInstituto de Química, Universidade de São Paulo, CP 780, 13560-970 São Carlos-SP, Brazil
ABSTRACT
The absorption spectra of DPH at fixed concentration do not change with water content in organic solvents. It exhibits monomer bands, such as those obtained in ethanol. The absorption did not change for solutions up to 54 and 46% of water in ethanol and DMSO, respectively, for [DPH] = 5.0 × 10-6 mol L-1 at 30 °C. However, at the same experimental conditions, a gradual sharp decay of the DPH fluorescence is observed. It is proposed that water molecules below these water concentration limits act as quenchers of the excited states of DPH. Stern-Volmer quenching constants by intensities measurements are 7.4 × 10-2 (water/ethanol) and 2.6 × 10-2 L mol-1 (water/DMSO). DPH lifetime measurements in the absence and presence of water resulted in 7.1 × 10-2 L mol-1 in water/ethanol, which pointed out that the process is a dynamic quenching by water molecules. For experiments using DPH as probe, this process can affect data, leading to misunderstanding interpretation.
Keywords: diphenylhexatriene, DPH, DPH fluorescence, dynamic quenching, lifetime
RESUMO
O espectro de absorção de DPH, em concentração fixa, não varia com o teor de água em solvente orgânico. Tem-se a banda de monômeros igual àquela em etanol puro. A absorção não muda até o limite de 54 e 46% de água em etanol e DMSO, respectivamente, para [DPH] = 5,0 × 10-6 mol L -1 a 30 °C. Entretanto, em misturas com água muito abaixo desses conteúdos críticos, observou-se um decaimento intenso de fluorescência enquanto a absorção manteve-se constante. Propõe-se que moléculas de água atuam como supressores dos estados excitados e a constante de supressão de Stern-Volmer através de intensidade relativas, resultou em 7,4 × 10-2 (água/etanol) e 2,6 × 10-2 L mol-1 (água/DMSO). Os tempos de vida do DPH na ausência e presença do supressor forneceram constantes de 7,1 × 10-2 L mol-1 em água/etanol, indicando supressão dinâmica. Em investigações de ambientes com esta sonda, este processo deve ser considerado tendo em vista o risco de erros de interpretação.
Introduction
trans-1,6-Diphenyl-1,3,5-hexatriene (DPH), which is used as a fluorescence probe, has a rod-like structure that absorbs and emits light with a high quantum efficiency, although it is not a large conjugative π-electron system.1,2 The spectrophotometric properties of DPH are very sensitive to the experimental conditions, which allows using this compound and its derivatives as environmental probes in colloidal systems to determine the critical micellar concentration of surfactants3 and in biological cell membranes.4,5 The measurement of the fluorescence anisotropy of DPH is extensively used for membranes, particularly in studies of microviscosities,6 due to the non-polar characteristic, the low water solubility, and the suitable geometric design, which maintains the molecule aligned parallel to the phospholipids chains of the membranes.7 Although the DPH molecule remains inserted in the membrane layer, it stays in contact with water molecules.2,5
The majority of DPH spectrophotometric studies on electronic transitions are performed in organic solvents.1,2,8-14

For DPH, the ground state (S0) and the first excited state (S1) have Ag symmetry, while the second excited state (S2) has Bu symmetry. Therefore, the So-S1 transition is not allowed by the symmetry rule, while the So-S2 transition is.10 Therefore, the DPH absorption band corresponds to the 1Ag-1Bu* (So-S2) transition. In contrast, the very long fluorescence lifetime of DPH indicates that the emission does not correspond to a reverse 1Bu*-1Ag (S2-So) transition. As known, the 1Ag* state (S1) is more stable than the 1Bu* state (S2) in long diphenylpolyenes, such as DPH. Birks11,12 first suggested that the low S1-S2 energy difference, about 1000 cm-1 in hexane, allows coupling (Herzberg-Teller vibronic coupling) of both states, as shown by the energy diagram in Scheme 1. This coupling changes the "purity" state of 1Ag* by orbital symmetry modifications, thus diminishing the forbiddance of DPH S1-So fluorescence emission after a quick non-radiative internal S2-S1 conversion.18 In fact, either fluorescence or rotation (cis-trans isomerization) occurs from 1Ag* (S1 state). As the coupling diminishes, the fluorescence yield, ΦF, decreases, while the cis-trans rotation increases.14
Several spectrophotometric and photochemical properties of DPH depend on environmental factors such as the temperature, solvent nature, etc. They influence the energy diagram, particularly the energy gap of S1-S2 states and their coupling, and accordingly, so are the characteristics of the involved orbital. Orbital Ag is assigned to a highly symmetric state, a "covalent" character, while the Bu state presents more "ionic" characteristics.10 Therefore, the energy of the 1Ag-1Bu* (So-S2 absorption) transition is affected by the solvent polarity and polarizability. A highly polar and polarizable solvent stabilizes the 1Bu* state (S2) better than the 1Ag state (So), which is less sensitive, leading to red-shifted absorption peaks. Dupuy and Montagu2 showed that the main absorptivity parameter of DPH is the solvent polarizability (αS) rather than the polarity, which is related to the refractive index. The higher the solvent refractive index, the greater the red-shifted absorption band.1,2 However, the solvent nature does not affect the fluorescence wavelength, since the (S1-So) process involves two Ag orbitals, which suffer the same type of stabilization by the solvent and are less sensitive to polarizability. Additionally, a solvent with high polarizability stabilizes the S2 orbital and favors the S2-S1 coupling, leading to a high fluorescence yield.9,15 The slightly decrease in the polarizability of organic solvents with the increase in the temperature destabilizes the 1Bu* state. The resulting decrease of the S1-S2 coupling promotes a decrease of ΦF.10,15,16 This effect is more evident in polar solvents, such as ethanol and acetonitrile, than in non-polar solvents.1 The present work investigated an unusual fluorescence behavior of DPH associated with the presence of water in organic solvents (ethanol and DMSO) attributed to DPH excited states quenching by water molecules.
Experimental
Anhydrous ethanol (Merck) and DMSO (Malinckrodt) were used as supplied. Sodium dodecyl sulfate (SDS) was purchased from Aldrich. Deionized and distilled water was used throughout. The DPH (all-trans, Aldrich) stock solutions were prepared in DMSO and kept frozen in the dark. Before use, the melted solutions were standardized by UV-Vis spectrophotometry in methanol (Merck) using a molar absorptivity of ε350 nm = 88000 L mol-1 cm-1.17 Approximately 100 mL of the solvent solution was prepared by mixing accurately measured volumes of water and organic solvents using a calibrated pipette. An aliquot of DPH stock solution was added to 2 mL of solvent mixture in a quartz cuvette using a micro syringe and agitated vigorously. The final samples contained no more than 0.07% (v/v) of DMSO and its presence did not influence the experimental results. All experiments were carried out at 30 °C. The absorption and fluorescence spectra were obtained in Varian Cary-50 and Perkin-Elmer LS-5 spectrometers, respectively. For fluorescence experiments, the excitation was fixed at 353 nm and the emission was monitored at 430 nm. For Resonance Light Scattering spectra the spectrofluorometer were operated in synchronous mode (Δλ = 0). The lifetime measurements were conducted in equipment from Optical Building Blocks Corporation (OBBC) with excitation source at 370 nm (LED) and emission at 435 nm, and the software EasyLife V. The fluorometer light exposure time was controlled to avoid DPH photodegradation.1 The amount of water is given as v/v percentage in organic solvent.
Results and Discussion
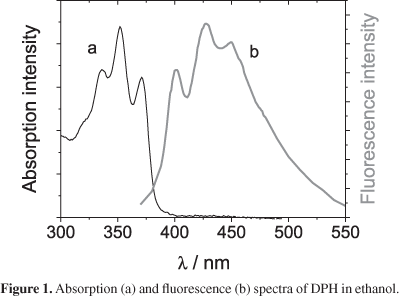
The electronic absorption spectrum of DPH in anhydrous organic solvents, such as ethanol, methanol, and DMSO, exhibits a long wavelength band (300-400 nm) composed of three characteristic vibrational peaks and shoulders that correspond to DPH in monomeric form. The absorption and fluorescence spectra of monomeric DPH are illustrated in Figure 1. The lack of a mirror image between these spectra is due to the electronic states involved once the absorption corresponds to So-S2* transition and the emission corresponds to S1*-So transition.12,13
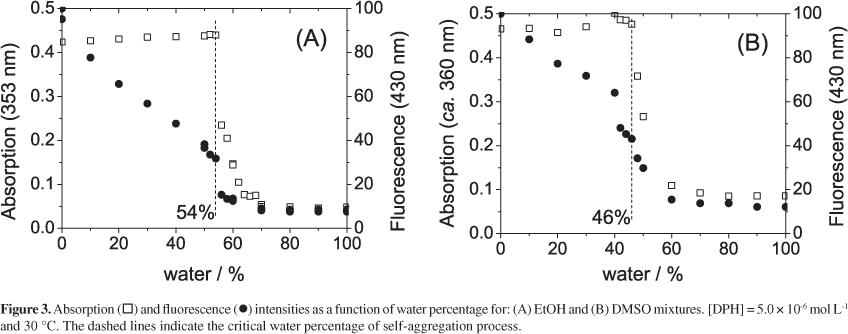
In the presence of large amounts of water in ethanol and in DMSO, the absorption spectra of DPH changed as illustrated in Figure 2 (water/ethanol system as an example). However, these changes for [DPH] = 5.0 × 10-6 mol L-1 at 30 °C are observed only above 54% water in ethanol and 46% water in DMSO.
These absorption spectral changes, expressed by a strong decrease in the intensity of the peak around 353 nm (the most intense monomer absorption peak) and a new absorption band at 300 nm, may be attributed to a DPH self-aggregation process that leads to dimers, trimers, and other aggregates. These results will not be discussed in the present report18 as its focus is presently on the experimental conditions below this critical amount of water and the resulting effects observed on the spectrophotometric properties of DPH. Therefore, all subsequent results are related to these water ranges, below 54% water in ethanol and below 46% in DMSO for [DPH] = 5.0 × 10-6 mol L-1 at 30 °C. In these ranges, while the absorption spectra did not vary, remaining almost the same as in anhydrous organic solvents, the fluorescence decays even for low water contents as illustrated in Figure 3. Indeed the absorbance at 353 nm presented a linear correlation with DPH concentration (Beer's plot) for [DPH] up to 5.0 × 10-6 mol L-1 in water/ethanol solution (experiment series at water ranging from 0 to 40%), not shown.
The absence of new absorption bands or peak shifts in the absorption and fluorescence spectra of DPH makes the isomerization hypothesis improbable, since cis-polyenes usually exhibit blue-shifted absorption bands when compared to trans-polyenes.19 This effect of water on the DPH fluorescence was previously reported by Dupuy and Montagu.2 In that work the fluorescence intensity decayed 16 and 29% in ethanol, and 3 and 13% in DMSO for 3 and 10% water, respectively. They assigned this phenomenon to DPH and water interactions without further discussion.5 While water does not shift the wavelength of the emission bands, its low polarizability, αS, water = 0.206, compared to 0.222 of ethanol and 0.284 of DMSO at 30 °C, destabilizes the S2 orbital and reduces the S2-S1 coupling, which are reflected as a weak fluorescence emission (low ΦF).9,15 However, the large decrease in the fluorescence intensity for such low water percentage in the solvent cannot be attributed to a simple solvent polarizability effect, as demonstrated by the fluorescence intensity decay of 27% for the addition of 10% water in methanol.2 The αS of methanol is even smaller than that of water (αS, methanol = 0.203). As the same time the observed fluorescence decrease can not be attributed to dielectric constant effects. Therefore, a specific mechanism of quenching process should exist, which promotes DPH fluorescence decreases. The fluorescence data was applied to the Stern-Volmer equation (1), considering water molecules acting as the quenchers:

where Fo is DPH fluorescence intensity in organic solvent, F is observed fluorescence intensity, KSV is Stern-Volmer quenching constant and [H2O] is water molar concentration. The plots of Fo/F against [H2O] for three fixed DPH concentrations are illustrated in Figure 4.
All plots in Figure 4 show linear dependence upon the quencher concentration. Table 1 shows the KSV values and the correlation coefficients (cc) of the fluorescence data. Figure 4 and the results in Table 1 show the excellent curve fitting and that the Stern-Volmer constants are independent of the DPH concentration. The average KSV value for the DPH in water/ethanol mixtures resulted in 7.4 × 10-2 L mol-1 while in water/DMSO 2.6 × 10-2 L mol-1 with cc = 0.9977. Therefore, it is demonstrated that the water molecules are acting as quenchers.
In the present work, two basic mechanisms: the dynamic (or collisional) quenching and the static quenching can be responsible for DPH deactivation. A similar ΦF decay in aqueous solutions of fluoresceins20,21 and cumarins22 was attributed to specific water molecule interactions caused by hydrogen bonding, as discussed by Arbeloa.20 Although hydrogen bonding or even polar-polar interactions are not recognizable for DPH, some authors have reported experimental and theoretical ground state benzene-water weak structures in which the aromatic rings would act as hydrogen bond acceptors.23 For DPH, as reported,10 the S2* electronic state (1Bu*) of DPH is assigned as more "ionic" symmetry and there is a S2*-S1* coupling that changes the "purity" of both states,12 which may turn the S1* state more "ionic", so that more sensible to interactions.
The fluorescence measurements at 30 and at 50 °C, represented as F30 and F50, respectively, showed a decrease in the intensity at higher temperatures for the water percentage range considered (F50/F30 ratio < 1). This effect was first explained by Alford and Palmer,16 who attributed it to a small decrease in polarizability with the rise in temperature, which destabilized the S2 orbital and resulted in the S2-S1 decoupling (decrease in ΦF). Additionally the temperature elevation favors internal conversion instead fluorescence emission. However, another explanation for the present case is the viscosity diminution as the temperature increases, resulting in faster diffusion and hence larger amounts of collisions (higher energy suppression), which suggests dynamic quenching. The value of the F50/F30 ratio was independent of the water content. Additionally, the fluorescence intensity decayed even with the addition of SDS to an aqueous ethanolic solution of DPH (not shown). These results eliminate the possibility of DPH self-aggregation for water contents below the previously mentioned critical water concentration, since the increase in temperature and the addition of surfactant cause monomer formation, which should result in higher fluorescence.24 In addition Resonance Light Scattering (RLS) experiments using the spectrofluorometer in synchronous mode did not show RLS signals that pointed out absence of aggregates in these solutions.
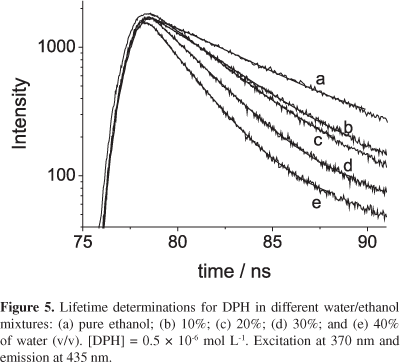
To confirm that the DPH fluorescence diminution by water molecules follows the mechanism of dynamic quenching, time-resolved experiments were performed at several water/ethanol conditions (Figure 5). From the data exposed in Figure 5, the fluorescence lifetimes of DPH in the absence (τo) and presence (τ) of quenchers were calculated. The obtained lifetime of DPH in pure ethanol (τo = 5.0 ns) is in accordance to the value found in the literature1 (5.0 ns at 32 °C in the absence of oxygen). Figure 6 presents the plot of τo/τ against water molar concentration.
As can be seen, the plot in Figure 6 follows a linear relationship whose slope is 7.1 × 10-2 L mol-1. This value is the same as the KSV obtained from fluorescence stead-state results (7.4 × 10-2 L mol-1). The equivalence of the ratios, Fo/F (Figure 4) and τo/τ (Figure 6), demonstrates that the investigated fluorescence decay is due to dynamic quenching of the DPH excited states by water molecules. Indeed, the calculated bimolecular quenching constant kqca. 1.5 × 107 L mol-1 s-1 (calculated from τo and KSV) is smaller than the typical diffusion-controlled value, which shows low quenching efficiency.
Conclusion
Once DPH is used as probe for colloid and membrane investigations, the large fluorescence decay of DPH in presence of water must be taken into account. Despite being hydrophobic, the DPH probe can be exposed to water molecules in some microenvironments. This quenching process can affect experimental data, leading to misunderstanding interpretation. The deactivation mechanism of excited states of DPH is due to the dynamic quenching by water molecules.
Acknowledgments
This work was supported by the Brazilian agencies: Fundação Araucária-Seti/Paraná, FAPESP, CNPq and CAPES NanoBiotec. The authors want to thank Laerte J. da Silva for the English language revision.
References
1. Cehelnik, E. D.; Cundall, R. B.; Lockwood, J. R.; Palmer, T. F.; J. Phys. Chem. 1975, 79, 1369.
2. Dupuy, B.; Montagu, M.; Analyst 1997, 122, 783.
3. Alexandridis, P.; Holzwarth, J. F.; Hatton, T. A.; Macromolecules 1994, 27, 2414; Hioka, N.; Chowdhary, R. K.; Chansarkar, N.; Delmarre, D.; Stenberg, E.; Dolphin, D.; Can. J. Chem. 2002, 80, 1321; Gaisford, S.; Beezer, A. E.; Mitchell, J. C.; Bell, P. C.; Fakorede, F.; Finnie, J. K.; Williams, S. J.; Int. J. Pharm. 1998, 174, 39.
4. Shinitzky, M.; Barenholz, Y.; J. Biol. Chem. 1974, 249, 2652.
5. Trotter, P. J.; Storch, J.; Biochim. Biophys. Acta, Biomembr. 1989, 982, 131.
6. Soto, M. A.; Sotomayor, C. P.; Lissi, E. A.; J. Photochem. Photobiol., A 2002, 152, 79; Lakowicz, J. R.; Principles of Fluorescence Spectroscopy, 2nd ed.; Plenum Press: New York, 1994.
7. Shinitzky, M.; Barenholz, Y.; Biochim. Biophys. Acta, Biomembr. 1978, 515, 367.
8. Turek, A. M.; Krishnamoorthy, G.; Sears, D. F.; Garcia, I.; Dmitrenko, O.; Saltiel, J.; J. Phys. Chem. A 2005, 109, 293; Hilinski, E. F.; McGowan, W. M.; Sears, D. F.; Saltiel, J.; J. Phys. Chem. 1996, 100, 3308.
9. Itoh, T.; Kohler, B. E.; J. Phys. Chem. 1987, 91, 1760.
10. Allen, M. T.; Whitten, D. G.; Chem. Rev. 1989, 89, 1691.
11. Birks, J. B.; Birch, D. J. S.; Chem. Phys. Lett. 1975, 31, 608; Birks, J. B.; Chem. Phys. Lett. 1978, 54, 430.
12. Birks, J. B.; Tripathi, G. N. R.; Lumb, M. D.; Chem. Phys. 1978, 33, 185.
13. Saltiel, J.; Wang, S.; J. Am. Chem. Soc. 1995, 117, 10761.
14. Saltiel, J.; Ko, D. H.; Fleming, S. A.; J. Am. Chem. Soc. 1994, 116, 4099.
15. Allen, M. T.; Miola, L.; Whitten, D. G.; J. Phys. Chem. 1987, 91, 6099.
16. Alford, P. C.; Palmer, T. F.; Chem. Phys. Lett. 1982, 86, 248.
17. Haugland, R. P.; Handbook of Fluorescent Probes and Research Chemicals, 6th ed.; Molecular Probes Inc.: New York, 1996.
18. Gracetto, A. C.; Batistela, V. R.; de Oliveira, H. P. M.; Hioka, N.; unpublished work.
19. Lunde, K.; Zechmeister, L.; J. Am. Chem. Soc. 1954, 76, 2308.
20. Arbeloa, F. L.; Arbeloa, T. L.; Bartolome, P. H.; Estevez, M. J. T.; Arbeloa, I. L.; Proc. Indian Acad. Sci. (Chem. Sci.) 1992, 104, 165.
21. Arbeloa, F. L.; Arbeloa, T. L.; Estevez, M. J. T.; Arbeloa, I. L.; J. Phys. Chem. 1991, 95, 2203; Arbeloa, F. L.; Arbeloa, T. L.; Arbeloa, I. L.; Costela, A.; Garcia-Moreno, I.; Figueira, J. M.; Amat-Guerri, F.; Sastre, R.; J. Lumin. 1997, 75, 309.
22. Arbeloa, F. L.; Arbeloa, T. L.; Arbeloa, I. L.; J. Lumin. 1996, 68, 149.
23. Suzuki, S.; Green, P. G.; Bumgarner, R. E.; Dasgupta, S.; Goddard, W. A.; Blake, G. A.; Science 1992, 257, 942; Li, S.; Cooper, V. R.; Thonhauser, T.; Puzder, A.; Langreth, D. C.; J. Phys. Chem. A 2008, 112, 9031.
24. Simplicio, F. I.; Maionchi, F.; Santin Filho, O.; Hioka, N.; J. Phys. Org. Chem. 2004, 17, 325; Simplicio, F. I.; Soares, R. R. S.; Maionchi, F.; Santin Filho, O.; Hioka, N.; J. Phys. Chem. A 2004, 108, 9384.
Received: January 18, 2010
Web Release Date: April 12, 2010
FAPESP has sponsored the publication of this article.
- 1. Cehelnik, E. D.; Cundall, R. B.; Lockwood, J. R.; Palmer, T. F.; J. Phys. Chem. 1975, 79, 1369.
- 2. Dupuy, B.; Montagu, M.; Analyst 1997, 122, 783.
- 3. Alexandridis, P.; Holzwarth, J. F.; Hatton, T. A.; Macromolecules 1994, 27, 2414;
- Hioka, N.; Chowdhary, R. K.; Chansarkar, N.; Delmarre, D.; Stenberg, E.; Dolphin, D.; Can. J. Chem. 2002, 80, 1321;
- Gaisford, S.; Beezer, A. E.; Mitchell, J. C.; Bell, P. C.; Fakorede, F.; Finnie, J. K.; Williams, S. J.; Int. J. Pharm. 1998, 174, 39.
- 4. Shinitzky, M.; Barenholz, Y.; J. Biol. Chem. 1974, 249, 2652.
- 5. Trotter, P. J.; Storch, J.; Biochim. Biophys. Acta, Biomembr. 1989, 982, 131.
- 6. Soto, M. A.; Sotomayor, C. P.; Lissi, E. A.; J. Photochem. Photobiol., A 2002, 152, 79;
- Lakowicz, J. R.; Principles of Fluorescence Spectroscopy, 2nd ed.; Plenum Press: New York, 1994.
- 7. Shinitzky, M.; Barenholz, Y.; Biochim. Biophys. Acta, Biomembr. 1978, 515, 367.
- 8. Turek, A. M.; Krishnamoorthy, G.; Sears, D. F.; Garcia, I.; Dmitrenko, O.; Saltiel, J.; J. Phys. Chem. A 2005, 109, 293;
- Hilinski, E. F.; McGowan, W. M.; Sears, D. F.; Saltiel, J.; J. Phys. Chem 1996, 100, 3308.
- 9. Itoh, T.; Kohler, B. E.; J. Phys. Chem. 1987, 91, 1760.
- 10. Allen, M. T.; Whitten, D. G.; Chem. Rev. 1989, 89, 1691.
- 11. Birks, J. B.; Birch, D. J. S.; Chem. Phys. Lett. 1975, 31, 608;
- Birks, J. B.; Chem. Phys. Lett. 1978, 54, 430.
- 12. Birks, J. B.; Tripathi, G. N. R.; Lumb, M. D.; Chem. Phys. 1978, 33, 185.
- 13. Saltiel, J.; Wang, S.; J. Am. Chem. Soc. 1995, 117, 10761.
- 14. Saltiel, J.; Ko, D. H.; Fleming, S. A.; J. Am. Chem. Soc. 1994, 116, 4099.
- 15. Allen, M. T.; Miola, L.; Whitten, D. G.; J. Phys. Chem. 1987, 91, 6099.
- 16. Alford, P. C.; Palmer, T. F.; Chem. Phys. Lett. 1982, 86, 248.
- 17. Haugland, R. P.; Handbook of Fluorescent Probes and Research Chemicals, 6th ed.; Molecular Probes Inc.: New York, 1996.
- 19. Lunde, K.; Zechmeister, L.; J. Am. Chem. Soc. 1954, 76, 2308.
- 20. Arbeloa, F. L.; Arbeloa, T. L.; Bartolome, P. H.; Estevez, M. J. T.; Arbeloa, I. L.; Proc. Indian Acad. Sci. (Chem. Sci.) 1992, 104, 165.
- 21. Arbeloa, F. L.; Arbeloa, T. L.; Estevez, M. J. T.; Arbeloa, I. L.; J. Phys. Chem. 1991, 95, 2203;
- Arbeloa, F. L.; Arbeloa, T. L.; Arbeloa, I. L.; Costela, A.; Garcia-Moreno, I.; Figueira, J. M.; Amat-Guerri, F.; Sastre, R.; J. Lumin. 1997, 75, 309.
- 22. Arbeloa, F. L.; Arbeloa, T. L.; Arbeloa, I. L.; J. Lumin. 1996, 68, 149.
- 23. Suzuki, S.; Green, P. G.; Bumgarner, R. E.; Dasgupta, S.; Goddard, W. A.; Blake, G. A.; Science 1992, 257, 942;
- Li, S.; Cooper, V. R.; Thonhauser, T.; Puzder, A.; Langreth, D. C.; J. Phys. Chem. A 2008, 112, 9031.
- 24. Simplicio, F. I.; Maionchi, F.; Santin Filho, O.; Hioka, N.; J. Phys. Org. Chem. 2004, 17, 325;
- Simplicio, F. I.; Soares, R. R. S.; Maionchi, F.; Santin Filho, O.; Hioka, N.; J. Phys. Chem. A 2004, 108, 9384.
Publication Dates
-
Publication in this collection
14 Oct 2011 -
Date of issue
2010
History
-
Received
18 Jan 2010