Abstracts
The anionic 5,10,15-tris(4-carboxyphenyl), 20-mono(2-nitrophenyl) porphyrin (1), 5,10(or 15)-bis(4-carboxyphenyl), 15(or 10),20-bis(2-nitrophenyl)porphyrin (2) and 5-mono(4-carboxyphenyl), 10,15,20-tris(2-nitrophenyl)porphyrin (3) were sinthesized directly by reaction of pyrrole with substituted benzaldehydes in nitrobenzene/propionic acid media. The benzaldehydes molar ratio was controlled to optimize the synthesis and purification of the desired porphyrins. This new series of porphyrins was characterised by TLC, mass spectrometry (FAB MS), ¹H NMR, UV/Vis, IR and electrochemistry. 5,10,15,20-Tetrakis(4-carboxyphenyl)porphyrin (4) and 5,10,15,20-Tetrakis(2-nitrophenyl)porphyrin (5) were also characterised for comparative purposes, completing the series The electrochemical reduction was investigated for the free base and corresponding iron(III) porphyrins on glassy carbon and mercury electrodes. The reduction potentials showed the expected dependence on the number of electron-withdrawing nitro groups present on the porphyrin ring providing additional evidences for the characterisation of the synthesised compounds.
anionic porphyrins; synthesis of porphyrins; metalloporphyrins; electrochemistry of porphyrins
As porfirinas aniônicas 5,10,15-tris(4-carboxifenil), 20-mono(2-nitrofenil) porfirina (1), 5,10(ou 15)-bis(4-carboxifenil), 15(or 10),20-bis(2-nitrofenil)porfirina (2) and 5-mono(4-carboxifenil), 10,15,20-tris(2-nitrofenil)porfirina (3), foram sintetizadas diretamente através da reação de pirrol com os benzaldeídos substituídos em meio de ácido propiônico/nitrobenzeno. A relação molar dos benzaldeídos foi controlada para otimizar a síntese e purificação das porfirinas desejadas. Esta nova série de porfirinas foi caracterizada por cromatografia em camada delgada, espectrometria de massas (FAB MS), RMN ¹H, UV/Vis, IV e eletroquímica. As porfirinas 5,10,15,20-tetrakis(4-carboxifenil)porfirina (4) e 5,10,15,20-tetrakis(2-nitrofenil)porfirina (5) também foram estudadas para comparação, tornando a série completa. A redução eletroquímica das porfirinas base livre e correspondentes ferro(III) porfirinas foi investigada em eletrodos de carbono e mercúrio. Os potenciais de redução mostraram a dependência esperada do número de grupos nitro, fortemente retirador de elétrons, presentes no anel porfirínico, fornecendo evidências adicionais para a caracterização dos compostos sintetizados.
Article
Synthesis and Characterization of a Novel Series of Meso (Nitrophenyl) and Meso (CarboxyPhenyl) Substituted Porphyrins
Marco A. Schiavona, Lidia S. Iwamoto * e-mail: mddassis@usp.br in memoriam a, Antônio G. Ferreirab, Yassuko Iamamotoa, Maria V. B. Zanonic and Marilda das D. Assisa* * e-mail: mddassis@usp.br in memoriam
aDepartamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto,
Universidade de São Paulo, Av. Bandeirantes, 3900, 14040-901, Ribeirão Preto - SP, Brazil.
bDepartamento de Química, Universidade Federal de São Carlos,
Rodovia Washington Luis Km 235, 13595-905, São Carlos - SP, Brazil.
c Instituto de Química, Universidade Estadual Paulista, CP 355, 14801-970, Araraquara - SP, Brazil
As porfirinas aniônicas 5,10,15-tris(4-carboxifenil), 20-mono(2-nitrofenil) porfirina (1), 5,10(ou 15)-bis(4-carboxifenil), 15(or 10),20-bis(2-nitrofenil)porfirina (2) and 5-mono(4-carboxifenil), 10,15,20-tris(2-nitrofenil)porfirina (3), foram sintetizadas diretamente através da reação de pirrol com os benzaldeídos substituídos em meio de ácido propiônico/nitrobenzeno. A relação molar dos benzaldeídos foi controlada para otimizar a síntese e purificação das porfirinas desejadas. Esta nova série de porfirinas foi caracterizada por cromatografia em camada delgada, espectrometria de massas (FAB MS), RMN 1H, UV/Vis, IV e eletroquímica. As porfirinas 5,10,15,20-tetrakis(4-carboxifenil)porfirina (4) e 5,10,15,20-tetrakis(2-nitrofenil)porfirina (5) também foram estudadas para comparação, tornando a série completa. A redução eletroquímica das porfirinas base livre e correspondentes ferro(III) porfirinas foi investigada em eletrodos de carbono e mercúrio. Os potenciais de redução mostraram a dependência esperada do número de grupos nitro, fortemente retirador de elétrons, presentes no anel porfirínico, fornecendo evidências adicionais para a caracterização dos compostos sintetizados.
The anionic 5,10,15-tris(4-carboxyphenyl), 20-mono(2-nitrophenyl) porphyrin (1), 5,10(or 15)-bis(4-carboxyphenyl), 15(or 10),20-bis(2-nitrophenyl)porphyrin (2) and 5-mono(4-carboxyphenyl), 10,15,20-tris(2-nitrophenyl)porphyrin (3) were sinthesized directly by reaction of pyrrole with substituted benzaldehydes in nitrobenzene/propionic acid media. The benzaldehydes molar ratio was controlled to optimize the synthesis and purification of the desired porphyrins. This new series of porphyrins was characterised by TLC, mass spectrometry (FAB MS), 1H NMR, UV/Vis, IR and electrochemistry. 5,10,15,20-Tetrakis(4-carboxyphenyl)porphyrin (4) and 5,10,15,20-Tetrakis(2-nitrophenyl)porphyrin (5)were also characterised for comparative purposes, completing the series The electrochemical reduction was investigated for the free base and corresponding iron(III) porphyrins on glassy carbon and mercury electrodes. The reduction potentials showed the expected dependence on the number of electron-withdrawing nitro groups present on the porphyrin ring providing additional evidences for the characterisation of the synthesised compounds.
Keywords: anionic porphyrins, synthesis of porphyrins, metalloporphyrins, electrochemistry of porphyrins
Introduction
The synthesis of porphyrins has gained special attention in recent years because of their importance in bioorganic and bioinorganic chemistry1 and their applications in biomedical sciences. Porphyrins are used, for example, to mimic the function of hemeproteins such as cytochrome P-450 in oxidation catalysis2, as photosensitizers in photodynamic therapy of cancer (PDT)3, in electron transport chains4 and as building blocks in molecular devices5. Each research area requires porphyrins with different and specific structural features, bearing a variety of different substituents.
Carboxy substituted porphyrins are attractive synthetic targets for three reasons: firstly, these substituents are present in natural porphyrins such as protoporphyrin IX, the prosthetic group of many important biological molecules like hemoglobin6. Secondly, the carboxy group confers an amphyphylic character to the porphyrins, and this is very important, for example, to improve the selectivity in tumour localization in PDT3. Lastly, carboxy groups can act as linkers to attach porphyrins to other materials, for example, they can be anchored to solid supports by amidation7.
We are interested in meso-para-carboxy and meso-ortho-nitro phenyl substituted porphyrins. The carboxy groups when ionised provide the charges necessary to enable their intercalation into lamellar double hydroxides (LDH)8 or to support them on cationic functionalized silicas through electrostatic binding. They can also be covalently linked to amino-functionalized silica to give heterogeneous porphyrin catalysts7. The ortho-nitro groups are important, since they remove electron density from the porphyrin ring and provide steric hindrance improving the stability of the metalloporphyrin catalysts in oxidation reactions9. The nitro group can improve the ability of porphyrin systems to act as radiosensitizers10 and, like carboxy groups, they can act as linkers to attach porphyrins to other materials7.
The purpose the present study was to combine the advantages offered by both functional groups into one porphyrin ligand. The corresponding metalloporphyrins will be used as catalysts for hydrocarbon oxidations in homogeneous and supported systems.
In this paper, we report the synthesis and purification of anionic tetraarylporphyrins bearing carboxy- and nitro- substituents on the phenyl rings (Figure 1), using the method of Gonsalves et al 11. The characterisation of water-soluble porphyrins is much more difficult than the characterisation of porphyrin derivatives soluble in organic solvents because of their salt-like character. UV/Vis and 1H NMR spectra are severely complicated by aggregation effects and a quantitative interpretation is rarely possible. The results of elemental analyses are not definitive because the substances are strongly hygroscopic12. Therefore, the synthesized porphyrins were characterised by combining thin layer chromato-graphy (TLC), mass spectrometry (FAB MS), 1H NMR, UV/Vis and IR spectroscopies and electrochemistry and by comparison with the commercial tetra-(para-carboxy-phenyl) and tetra-(ortho-nitrophenyl) porphyrins.
Experimental
Materials and methods
Commercially available chemicals and solvents were from Aldrich, Fisons, Sigma and Merck, unless otherwise stated. Silica gel (230-400 mesh) was purchased from Merck and Florisil (magnesia silicate 60-100 mesh) from J.T. Baker. 5,10,15,20-Tetrakis(4-carboxyphenyl)porphyrin (4) and 5,10,15,20- tetrakis(2-nitrophenyl)porphyrin (5) and 5,10,15,20-tetraphenylporphyrin (6) were purchased from Midcentury and used as received.
UV/Vis spectra were recorded on a Hewlett Packard 8452 Diode Array spectrophotometer. The spectra were recorded in 2 mm path length quartz cells (Hellma). The FTIR spectra were recorded on a FT spectrometer-BOMEN B-100 (4000 - 400 cm-1) in KBr pellets; the FeP : KBr molar ratio was aproximately 1:2000. FAB mass spectra were recorded on a V.G. Analytical Autospec Instrument, using FAB+ mode. 4-Nitrobenzyl alcohol was used as the matrix and caesium ion bombardment to generate the ions. The 1H NMR measurements were recorded on a BRUKER 400.13 MHz, DRX400 spectrometer operating at 303 K using a 5 mm BBO probe with temperature control. The 1H NMR spectra of 2, 3 and 5 were obtained in CD2Cl2 solution (2,5 x 10-2 mol dm-3) and for 1 and 4 CD3OD solutions of the same concentration were used. All spectra were acquired with spectral widths of approximately 16000 Hz and 64 K data points for acquisition using TMS as internal reference. All electrochemical experiments were carried out with a Metrohm Polarecord E-506 coupled to a Metrohm 663 VA Stand which was connected to a computer for data storage and handling. A multimode electrode system was used in both the dropping mercury electrode for differential pulse polarographic measurements and the glassy carbon electrode for cyclic voltammetric measurements. The glassy carbon electrode was polished with alumina before each experiment, rinsed with water, cleaned in a ultrasound bath and dried at room temperature. The three-electrode system was completed with a glassy carbon auxiliary electrode and an Ag/AgCl (3 mol dm-3 KCl) reference electrode. A pulse amplitude of 50 mV was used for differential pulse polarography. Tetra-n-butyl-ammonium tetraphenylborate (TBATFB) was obtained from Eastman Chemicals and was used as received. Porphyrin solutions were 1x10-4 mol dm-3, prepared by dissolving the purified substance in CH2Cl2.
Synthesis and purification of porphyrins and metalloporphyrins
5,10,15-Tris(4-carboxyphenyl)20-mono(2-nitrophenyl) porphyrin (1), 5,10(or 15)-bis(4-carboxyphe-nyl)15(or 10),20-bis(2-nitrophenyl) porphyrin (cis- or trans-isomer) (2) and 5-mono(4-carboxyphenyl)10,15,20-tris(2-nitrophenyl) porphyrin (3) were prepared according to the method of Gonsalves et al11. The procedure was repeated with different ratios of 2-nitrobenzaldehyde to 4-carboxybenzaldehyde in order to optimize the yields of the desired porphyrin.
Synthesis of 1
2-Nitrobenzaldehyde (1.47 g, 9.8 mmol) and 4-carboxybenzaldehyde (3.04 g, 20.2 mmol) were mixed with propionic acid (105 cm3) and nitrobenzene (45 cm3) and pyrrole (2.08 cm3, 30 mmol) was added. The reaction vessel was shielded from ambient light and the mixture was heated at 120 °C for 1 h. After cooling and solvent removal under vacuum the crude porphyrin was adsorbed onto 10.5 g of Florisil. This mixture was applied to a dry-packed Florisil column and eluted with solvents of increasing polarity: dichloromethane, dichloromethane/methanol (9 : 1), dichloromethane/methanol (1 : 1), and methanol. The last two fractions which contained porphyrins 1 and 2, were filtered to eliminate the Florisil particles. The solvent was removed under vacuum and the solid porphyrins were finally purified by column chromatography on silica with dichloromethane/acetone/acetic acid (8 : 2 : 0.1) as eluent to give 297 mg (3.75 x 10-4 mol, 5% yield) of pure 1, lmax/nm (CH3OH) 416 (e/dm3 mol-1 cm-1 2.3 x 105), 514 (1.3 x 104), 546 (6.9 x 103), 590 (5.7 x 103), 644 (3.7 x 103); m/z (M+) 792.
Synthesis of 2
This compound was obtained as a by-product from the syntheses of 1 and 3, lmax/nm (CH3OH) 416 (e/dm3 mol-1 cm-1 2.1 x 105), 514 (1.3 x 104), 548 (5.5 x 103), 590 (4.0 x 103), 648 (2.2 x 103); m/z (M+) 793.
Synthesis of 3
2-Nitrobenzaldehyde (1.81 g, 12 mmol) and 4-carboxy-benzaldehyde (0.45 g, 3 mmol) were mixed with propionic acid (52.5 cm3) and nitrobenzene (22.5 cm3) and pyrrole (1.04 cm3, 15 mmol) was added. The reaction and purification were similar for 1 giving 148 mg (1.86 x 10-4 mol, 5 % yield) of pure 3, lmax /nm (CH3OH) 418 (e/dm3 mol-1 cm-1 1.0 x 105), 514 (8.7 x 103), 550 (4.6 x 103), 590 (4.0 x 103), 650 (1.9 x 103); m/z (M+) 794.
Synthesis of iron(III)porphyrins, FeP (1-Fe, 2-Fe, 3-Fe, 4-Fe, 5-Fe)
These compounds were prepared by refluxing the free base porphyrins (amounts corresponding to 0.2 mmol) with iron(II) chloride tetrahydrate (amount corresponding to 2 mmol) in N,N'-dimethylformamide (15 cm3) under nitrogen by the method of Adler et al13. The reaction was monitored by UV/Vis spectroscopy and TLC. The iron porphyrins were precipitated from the reaction medium by adding HCl 0.10 mol dm-3 (15 cm3), and cooling the reaction flask in an ice bath. The precipitates were recovered by centrifugation and the supernatant, which contained DMF and excess iron(II) salt, was discarded. The iron porphyrins were washed with HCl 0.10 mol dm-3 (30 cm3) and purified by silica column chromatography. The solvents used to elute firstly the free base porphyrin followed by the iron porphyrins were: methanol and methanol/acetic acid mixture (10:0.1) for 1-Fe; a dichloromethane/acetone/acetic acid mixture (8 : 2: 0,1) for 2-Fe; dichloromethane and a dichloromethane/methanol mixture (9:1) for 3-Fe. These procedures gave the amounts corresponding to metallation yields of 1-Fe - 64%; lmax/nm (CH3OH) 338 (e/dm3 mol-1 cm-1 2.2 x 104), 416 (6.2 x 104), 582 (3.5 x 103), 656 (shoulder). m/z (M+) : 845. 2-Fe - 55%; lmax/nm (dichloroethane) 372 (e/dm3 mol-1 cm-1 3.2 x 104), 422 (6.2 x 104), 510 (1.0 x 104), 582 (4.5 x 103), 670 (broad, 3 x 103). m/z (M+): 846; 3-Fe - 72%; lmax/nm (dichloroethane) 368 (e/dm3 mol-1 cm-1 3.8 x 104), 422 (7.2 x 104), 510 (1.1 x 104), 582 (5.4 x 103); m/z (M+) 847.
Results and Discussion
Porphyrins syntheses
The greatest advance in the development of methods for porphyrin synthesis since the classic Rothemund process14 has been the procedure of Lindsey et al15.The gentle conditions of this synthesis provide means for converting a large range of pre-functionalized benzal-dehydes into the corresponding porphyrins. For this reason, the method has been widely employed over recent years. However, Lindsey's method fails when the porphyrin synthesis involves benzaldehydes bearing ionic substituents which are insoluble in the reaction solvents dichloromethane or chloroform, unless the ionic group is masked. In this way, the synthesis of porphyrins bearing carboxy groups often requires the acid groups to be masked as esters. This consequently involves an extra step of group deprotection after the porphyrin has been formed16. We decided to use the method of Gonsalves et al11, which employs propionic acid and nitrobenzene medium, in the synthesis of the ortho-nitrophenyl and para-carboxyphenyl substituted porphyrin series. Whereas in Lindsey's method an expensive highly potent quinone is used as the oxidant, in the method of Gonsalves the oxidant is the nitrobenzene/propionic acid solvent. The latter also leads to the precipitation of the porphyrin from the reaction medium, thus facilitating its isolation and purification.
A mixed aldehyde condensation using stoichiometric amounts of ortho-substituted benzaldehyde (A), para-substituted benzaldehyde (B) and pyrrole affords a mixture of six different porphyrins which can be separated chromatographically5a. In order to decrease the number of porphyrins obtained in the synthesis and to avoid extremely tedious chromatographic purification processes the system was optimized with small scale reactions using different ratios of the two aldehydes. In this way each of the porphyrins A3B, A2B2 (cis and trans) and AB3 were prepared.
In the synthesis of 1 (AB3) porphyrin, the initial ratio of pyrrole:ortho-nitrobenzaldehyde:para-carboxybenzal -dehyde used was 4:0.8:3.2, rather than the stoichiometric 4:1:3 ratio. This synthesis gave a crude yield of 36% (estimated by the absorption of the Soret band in the UV/Vis spectrum of the reaction mixture) of the two porphyrins 4 and 1 at a 13:1 ratio as determined through the mass of porphyrins after purification. In a second experiment, taking into consideration the higher reactivity of para-carboxybenzaldehyde, the relative amount of ortho-nitrobenzaldehyde was increased and the reagents ratio used was 4:1.6:2.4. This synthesis led to a high crude yield (60% estimated by UV/Vis) of the mixture of the three porphyrins 1, 2 and 3. In a third experiment, the relative amounts of the two benzaldehydes (4:1.3:2.7) were fitted to obtain a high crude porphyrin yield (60%, estimated by UV/Vis) corresponding to the mixture of 1 and 2. This was the optimized condition used to obtain 1. The final yield of 1 after purification was 5%, based on pyrrole.
To synthesize the (A3B) porphyrin 3, we used pyrrole : o-nitrobenzaldehyde : p-carboxybenzaldehyde at a molar ratio of 4 : 3.2 : 0.8. In this synthesis, the porphyrins 5, 3 and 2 were detected and the desired porphyrin 3 was obtained in 5% yield based on the starting pyrrole, after purification. Such yields are good, considering the losses in purification and if compared to other similar preparations involving mixed substituents3b,3c,17,18.
For porphyrins purification, a careful study of the total polarity of the eluent to be employed was necessary. This study was carried out using TLC on silica gel and the ternary solvent mixtures initially used, such as benzene/methanol/acetic acid, water/acetonitrile/p-dioxane or 2,6-lutidine/water/ammonia (gas) described in the literature for natural and synthetic systems containing different numbers of carboxylic groups19. However, these water/solvent mixtures were not effective in the present study. Winkelman's20 quaternary solvent system, pyridine/chloroform/water/acetic acid (2:1:1:1) employed for the separation of porphyrins containing different numbers of SO3H groups, obtained from the sulfonation of 4, was also ineffective for separating the o-nitro- and p-carboxy-substituted porphyrins. Finally, by using the ternary solvent mixture, chloroform/acetone/acetic acid (8 : 2 : 0,1) proposed by Harada et al21 for the separation of the meso-mono-(p-carboxyphenyl)tri-phenylporphyrin from the mixture of this porphyrin and meso-tetraphenyl-porphyrin, good separations were obtained. By substituting chloroform for dichloromethane, we obtained the ideal solvent mixture for the purification of the porphyrins to be studied. Porphyrins 4 and 5 were also analysed in this solvent mixture as references. A difference of 0.10 0.20 in the Rf values per added carboxy group was observed (see Table I), with the exception of 4, which did not elute from the origin. The fraction containing 2 was probably a mixture of the cis and trans isomers. However it was not possible to separate them under the conditions used.
In conclusion the mixed ortho-nitrophenyl and para-carboxyphenyl substituted porphyrins can be obtained directly by the Gonsalves method. Furthermore, by controlling the molar ratio of the benzaldehydes, it is possible to optimize the synthesis and purification of the desired porphyrins.
Infrared spectroscopy
Infrared spectroscopy was very useful for the characterisation of the studied porphyrin series, due to the presence of the NO2 and COOH substituents, whose absorption bands are defined and easy to assign.
Compounds containing the NO2 group have strong absorption bands from symmetric and asymmetric deformations (1389 - 1259 cm-1 and 1661 - 1499 cm-1, respectively)22. The exact position of these bands depends on the substitutions and unsaturations within the NO2 group. Collman et al23 assigned the 1542-1526 cm-1 and 1356-1340 cm-1 absorption ranges to the asymmetrical and symmetrical vibrations, respectively, for the NO2 groups in 5,10,15,20-tetrakis-(o-nitrophenyl)porphyrin), 5. Goldberg et al24 reported these absorptions at 1500 and 1330 cm-1, respectively, for the same porphyrin. Table 2 shows the absorption frequencies observed for these groups on the porphyrins synthesized in this study. The values for 4 and 5 are also included for comparison purposes.
Porphyrins containing NO2 groups as substituents on the meso-phenyl rings (Table 2) show the characteristic absorption bands of this group which agree with Collman's results23. The relative absorption intensities decrease in the order 5, 3, 2 and 1, as would be expected upon decreasing the number of NO2 groups in these porphyrins (Figure 2).
The carboxylic acids are well assigned in FTIR spectroscopy to regions near 1700, 1400 and 1250 cm-1 of the spectrum22 and the study of all these regions provide a reliable identification of these acids. The absorption near 1700 cm-1 is assigned to C=O stretching vibration. Carboxylic acids are capable of forming hydrogen bonds between the carbonyl and hydroxyl groups of the two molecules and this carbonyl frequency is accordingly reduced25. In this study, the porphyrins bearing carboxy substituents show carbonyl frequencies near 1690 cm-1 (Table 2 and Figure 2) indicating aggregation through hydrogen bonds.
The two other absorptions, near 1400 cm-1 (1440 1395 cm-1) and near 1250 cm-1 (1320 1210 cm-1) have been associated by many workers to the single bond C-O stretching vibration25. These bands appear in the spectrum of 4 as very intense and large bands centred at 1402 and 1268 cm-1, which agree with data reported by Datta-Gupta and Bardos for this free-base porphyrin16. The intensity of these bands decreases in the order: 4 > 1 > 2 > 3, as expected, due to the decrease in the number of carboxylic acid groups in these porphyrins (Figure 2).
The others bands, not assigned here, are atributed to the vibration of the ring porphyrinic and phenyls group meso-substituents, and are well established in the literature16,26.
1H NMR spectroscopy
The high frequency region of the 1H NMR spectra shows the chemical shifts for pyrrole hydrogens (Hb) and these data were used to characterise the porphyrins. Table 3 shows the chemical shifts and the multiplicities of the signals .
The chemical shifts (d 8.69 br s) for Hb hydrogens (close to the nitrophenyl groups) of 5, are at lower frequency than the Hb hydrogens (close to the carboxyphenyl groups) of 4 (d 8.88 br s). This effect is caused by the electron withdrawing effect of the ortho-nitro groups on the phenyl ring, which leads to less deprotection by the anisotropy effect on these b-pyrrole hydrogens.
For 1, one signal (multiplet, d 8.78 8.83) was observed for Hb hydrogens between the carboxyphenyl substituents (Hc, Figure 3) and two signals (doublets) for the other Hb hydrogens. The latter arise from hydrogens Ha, which are close to the nitrophenyl group (d 8.69); and the former from Hb (d 8.80) next to the carboxyphenyl group (Figure 3). The chemical shift differences for hydrogens Ha and Hb reflect the difference in the electron current ring, which induces different anisotropic effects on them. However, for hydrogens Hc, we expected to see a singlet and not a multiplet27. This is probably because the molecular aggregation leads to a non-equivalency between hydrogens Hc. Unfortunately, the N-H pyrrole hydrogens were not observed for this compound because the NMR spectrum was obtained in methanol d-4 since this porphyrin is insoluble in dichloromethane. Integration of the areas corresponding to Hb (close to carboxy) and Hb (close to nitro) hydrogens of 3, 2 and 1 gave the expected 3:1, 2:2 and 1:3 (Hb-NO2/Hb-COOH) ratios, respectively. For 2, 1 and 4 we can also observe a broad hump above the signal corresponding to hydrogens Hb (close to nitro) and Hb (close to carboxy), which we again suppose are due to intermolecular aggregation. It is well known that porphyrins bearing carboxylic groups can aggregate through hydrogen bonds and also the polar nitro substituent can act as a proton acceptor in hydrogen bonds24. The aggregation was indicated also by infrared spectra, which show broad signals corresponding to the carboxyl group.
The chemical shifts of the N-H hydrogens in the centre of the porphyrin are also affected by the number of nitrophenyl groups. Increasing the number of nitro groups leads to the expected high frequency shift of the N-H signal. In the region of the phenyl hydrogens the multiplet signals overlap makes the assignments difficult.
Electrochemical characterizacion
The electrochemical reduction of free base and iron ortho nitro and para-carboxy substituted porphyrins was studied both on a glassy carbon electrode and a mercury electrode by cyclic voltammetry and differential pulse polarography, respectively, in CH2Cl2; 0.1 mol dm-3 tetra-n-butylammonium tetraphenylborate (TBATFB).
The electroreduction of the free bases 5,10,15,20-(tetraphenyl)porphyrin (6) and tetrakis(o-nitrophenyl) porphyrin (5) and the corresponding iron porphyrins, 6-Fe, 5-Fe, were also investigated in order to understand the influence of substituents on the electrochemical behaviour of these compounds and for comparison purposes. The free base porphyrins 1, 4 and corresponding iron porphyrins were not investigated by this technique due to their insolubility in dichloromethane which was used as solvent in these studies.
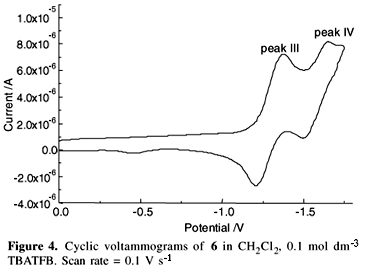
The voltammetric reduction of 6 has two characteristic reversible one-electron transfer reactions (Figure 4, Table 4) as judged by wave analysis of ip/n1/2 = constant, Epa-Epc = 59 mV and ipa/ipc = 0.9-1.0 for n (scan rate) = 10-2000 mV s-1. These reductions correspond to the production of p-anion and di-anion radicals as well established in literature28. The cyclic voltammograms for nitro-substituted compounds, 2 (Figure 5), 3 and 5, exhibit two reduction processes at very close potentials (Table 4). Only one anodic peak associated with the reoxidation of product generated in the second reduction step is seen on the reverse scan, but the ipa/ipc ratio is always smaller than one. In addition, both peak potentials show cathodic shifting as a function of scan rate increasing, indicating that nitro-substituted compounds follow a reduction mechanism involving chemical reactions subsequent to the electron transfer29. In general, all nitro-substituted free base porphyrins are easier to reduce relative to the parent unsubstituted porphyrin (see Table 4).
A comparison of the electrochemical behaviour with other reported data30,31 leads us to suppose that the redox reactions of the nitro-porphyrins reported here also involve the porphyrin p-ring system. Considering the predominant effect of the nitro groups on the electrochemical properties, all results were analysed as a function of the number of these groups.
Electrochemical characterization
In order to confirm these results, differential pulse polarographic experiments were carried out with the aim to improve wave separation. The same trends are observed in the polarographic behaviour of these compounds, as shown in Table 5. Differential pulse polarograms show well defined reduction waves and the corresponding peak potentials indicate that the reductions are easier, increasing the number of nitro groups on the porphyrin ring. Although the electrochemical behaviour in both electrode systems was similar, a negative shifting in the reduction steps was noticed as compared to that obtained with a glassy carbon electrode, indicating that the nature of the electrode material affects the reduction process.
A typical cyclic voltammogram obtained for nitro substituted iron(III) porphyrins is shown in Figure 6 and compared to that for 6-Fe obtained under the same conditions (Figure 7). All potential values obtained from voltammetric reduction of the iron porphyrins are shown in Table 4. As expected, the compounds containing the metal centre show two extra peaks at less negative potentials than that observed for reduction of the free base porphyrins. These peaks were assigned to the successive reduction of the iron (III)-iron(II) and iron (II)-iron (I) couples in the porphyrin complex32,33. However, the cyclic voltammograms are significantly changed in morphology and potential magnitude as the number of nitro groups is increased in the macrocycle.
All investigated FeP undergo a reduction process involving a reversible one electron transfer of the metal centre to yield Fe(II), as demonstrated by analysis of the scan rate influence, which shows typical virtually constant values of the ip/n1/2 ratio and Epa-Epc values always around 60 mV. Concomitantly, the peak potential shifts to less negative potential for FeP upon going from 6-Fe to 2-Fe, 3-Fe and then to 5-Fe, increasing the number of nitro groups in the macrocycle. The results are confirmed by polarographic techniques, as shown by peak potentials obtained at the mercury electrode (Table 5).
The metal centre second reduction process in the 6-Fe also shows a characteristic reversible reduction involving one electron transfer. Nevertheless, the three nitro derivatives investigated exhibit cyclic voltammograms with the second reduction peak at less negative potential than that required for reduction of the iron(II)-iron(I) couple in 6-Fe. The difference between the first and second reduction potentials decreases as the number of nitro substituents increases. These data may indicate that the second reduction potential is markedly more sensitive to changes in the electronic nature of the porphyrin macrocycle. The cathodic shift of the peak potential could be interpreted as an increased interaction of the iron(II) with the porphyrin ring. The absence of an anodic peak on the reverse potential scan at a slow scan rate indicates that the second reduction process is complicated by fast chemical reactions which probably consume the generated product, assigned as the iron(I) porphyrin complex, under these conditions. This chemical reaction could be the loss or exchange of the axial ligand34 or self protonation of the porphyrin35.
In addition, the reduction potential of the porphyrin ring system for all iron complexes is shifted up to 690 mV with the introduction of nitro groups, as shown in Tables 4 and 5. As previously observed, the reduction of free base nitro-porphyrins or corresponding iron porphyrins showed the same general trends, indicating that the ring system reduction strongly depends on the number of nitro substituents in the porphyrin macrocyle.
In conclusion, our findings suggest that the electronwithdrawing NO2 groups decrease the electron density in both metal centre and conjugated porphyrin p-ring system and this leads to easier reduction. On the other hand, the stability of the product electrochemically generated by the reduction of the Fe(II)-Fe(I) couple is decreased for the nitro-substituted porphyrins investigated in this study. Further work is now in progress in our laboratory in order to clarify the reduction mechanism of these compounds.
Conclusions
Despite great difficulties in working with ionic porphyrins due to aggregation, we have synthesized, isolated and characterised a new series of porphyrins containing mixed substituents, nitro and carboxy, in the meso-phenyl rings, with estimated overall porphyrin yield as high as 60%. We have demonstrated that it is possible to optimise the benzaldehyde molar ratio in order to obtain fewer porphyrin isomers and higher concentrations of the desired porphyrins, which facilitates the purification process. These porphyrins are important as possible precursors of systems of the self-assembly type and they are potentially good catalysts due to the versatility of the carboxy and nitro groups which can be used to support them in different materials. Further studies are currently under way to explore the catalytic activities of these compounds in oxidation reactions.
Acknowledgements
This work was supported by CAPES, CNPq and FAPESP. We thank J. R. Lindsay Smith for discussions and for the FAB MS spectra.
References
1. (a) Lindsey, J. S. Metalloporphyrins-Catalyzed Oxidations, Montanari, F. and Casella, L. Eds., Academic Publishers, The Netherlands, 1994. (b) Milgron, L.R. The Colours of Life, Oxford University Press, New York, 1997.
2. (a) Ortiz de Montellano, P. R. Cytochrome P450: Structure, Mechanism and Biochemistry, 2nd ed., Plenum Press, New York, 1995. (b) Dolphin, D.; Traylor, T.G.; Xie, L. Y. Acc. Chem. Res.1997, 30, 251.
3. (a) Bonnett, R. Chem. Soc. Rev. 1995, 19. (b) Gaud, O.; Granet, R.; Kaouadji, M.; Krausz, P.; Blais, J. C.; Bolbach, G. Can. J. Chem. 1996, 74, 481. (c) Driaf, K.; Granet, R.; Krausz, P.; Kaouadji, M.; Thomasson, F.; Chulia, A. J.; Verneuil, B.; Spiro, M.; Blais, J. C.; Bolbach, G. Can. J. Chem. 1996, 74, 1550.
4. Arimura, T. J. Synthetic Org. Chem. Japan 1997, 55, 557.
5. (a) Lindsey, J. S.; Prathapan, S.; Johnson, J. E.; Wagner, R. W. Tetrahedron 1994, 50, 8941. (b) Drain, C. M.; Nifiatis, F.; Vasenko, A.; Batteas, J. D. Angew. Chem. Int. Ed. 1998, 37, 2344. (c) Vicente, M. G. H.; Cancilla, M. T.; Lebrilla, C. B.; Smith, K. M. Chem. Commun. 1998, 2355. (d) Biemans, H. A. M.; Rowan, A. E.; Verhoeven, A.; Vanoppen, P.; Latterini, L.; Schenning, J.; Meijer, E. W.; Schryver, F. C.; Nolte, R. J. M. J. Am. Chem. Soc. 1998, 120, 11054. (e) Osuka, A.; Shimidzu, H. Angew. Chem. Int. Ed. Engl. 1997, 36, 135.
6. Kaim, W.; Schwederski, B. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life , John Wiley & Sons, Chichester, 1995.
7. Lindsay Smith, J. R. Metalloporphyrins in Catalytic Oxidations, Sheldon, R. A. Ed., Marcel Dekker, New York, 1994.
8. (a) Bedioui, F. Coord. Chem. Rev. 1995, 144, 39. (b) Mansuy, D.; Battioni, P.; Battioni, J. P. Eur. J. Biochem. 1989, 184, 267.
9. (a) Assis, M. D.; Serra, O. A.; Iamamoto, Y. Inorg. Chim. Acta 1991, 187, 109. (b) Assis, M. D.; Mello, A. J. B.; Serra, O. A.; Iamamoto, Y. J. Mol. Catal A: Chemical 1995, 97, 41.
10. Meng, G. G.; James, B. R.; Skov, K. A. Can. J. Chem. 1994, 72, 1894, and references therein.
11. Rocha Gonsalves, A. M. d'A.;Varejão, J. M. T. B.; Pereira, M. M. J. Heterocyclic Chem. 1991, 28, 635.
12. (a) Buchler, J. W.; Kunzel, F. M.; Mayer, U.; Nawra, M. Fresenius J. Anal. Chem. 1994, 348, 371. (b) Herrmann, O.; Medhi, H.; Corsini, A. Can. J. Chem. 1978, 56, 1084.
13. Adler, A. D.; Longo, F. R.; Kampas, F.; Kim, J. J. Inorg. Nucl. Chem. 1970, 32, 2445.
14. (a) Rothemund, P. J. Am. Chem. Soc.1936, 58, 625. (b) Rothemund, P. J. Am. Chem. Soc. 1939 61, 2912.
15. (a) Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827. (b) Lindsey, J. S.; Wagner, R. W. J. Org. Chem. 1989, 54, 828.
16. Datta-Gupta, N.; Bardos, T. J. J. Heterocyclic Chem. 1966, 3, 495.
17. (a) Bigey, P.; Frau, S.; Lomp, C.; Claparols, C.; Bernadou, J.; Meunier, B. Bull. Soc. Chim. Fr.1996, 13, 679. (b) Assis, M. D.; Lindsay Smith, J. R. J. Chem. Soc., Perkin Trans.II 1998, 2221.
18. Bonar-Law, R. P. J. Org. Chem. 1996, 61, 3623.
19. (a) Grishima, L. E.; Brykina, G. D.; Shpigum, O. A. J. Anal. Chem. 1995, 50, 826. (b) Jensen, J. J. Chromatog. 1963, 10, 236. (c) Chu, T. C. and Chu, E. J. J. Chromatog. 1966, 21, 46.
20. (a) Wilkelman, J. Cancer Research, 1962, 22, 589. (b) J. Wilkeman, J.; Slater, G.; Grossman, J. Cancer Research 1967, 27, 2060.
21. Harada, A.; Shiotsuki, K.; Yamaguchi, H.; Kamachi, M. Inorg. Chem. 1995, 34, 1070.
22. Silverstain, R. M.; Bassier, G. C.; Morril, T. C. Spectrometric Identification of Organic Compounds, John Wiley and Sons, Inc., 3a ed., 1974 .
23. Collman, J. P.; Brauman, J. I.; Doxee, K. M.; Halbert, T. R.; Bunnenberg, E.; Linder, R. E.; Lanar, G. N.; Gaudio, J. D.; Spartalian, K. J. Am. Chem. Soc. 1980, 102, 4182.
24. Kumar, R. K.; Balasubramanian, S.; Goldberg, I. Inorg. Chem. 1998, 37, 541.
25. Bellamy, L. J. The Infrared Spectra of Complex Molecules, John Willey and Sons, New York, 1954, ch. 10.
26. Thomas, D. W.; Martell, A. B. J. Am. Chem. Soc. 1959, 81, 5111.
27. Meng, G. G.; James, B. R.; Skov, K. A. Can. J. Chem. 1994, 72, 1894.
28. Fuhrlop, J. H.; Kadish, K. M.; Davis, D. G. J. Am. Chem. Soc. 1982, 95, 5140.
29. Bard, A. J. Electrochemical Methods, John Willey and Sons, New York,1980.
30. Giraudeau, A.; Callot, H. J.; Gross, M. Inorg. Chem. 1979, 18, 201.
31. Arao, T.; Maiya, B. G. Polyhedron 1994, 13,1863.
32. Messet, M. J. M.; Shokhirev, N. V.; Enemark, P. D.; Jacobson, S. E.; Walker, F. A. Inorg. Chem. 1996, 35, 5188.
33. Kadish, K. M.; Morrison, M. M.; Constant, L. A.; Dickens, L.; Davis, D. G. J. Am. Chem. Soc.1976, 98, 8337.
34. (a) Bottomley, L. A.; Kadish, K. Inorg. Chem. 1981, 20, 1348. (b) Lexa, D.; Momenteau, M.; Mispeltier, J.; Lhoste, J.M., Bioelectrochem. Bioenerg. 1975, 1, 108.
35. (a) Davis, D. G. in "The Porphyrins", D. Dolphin Ed., v.V, ch. 4, 19. (b) Constant, L. A.; Davis, D. G. Anal. Chem. 1975, 47, 2253.
Received: May 31, 1999
Published on the web: August 31, 2000
FAPESP helped in meeting the publication costs of this article.
- 1. (a) Lindsey, J. S. Metalloporphyrins-Catalyzed Oxidations, Montanari, F. and Casella, L. Eds., Academic Publishers, The Netherlands, 1994.
- (b) Milgron, L.R. The Colours of Life, Oxford University Press, New York, 1997.
- 2. (a) Ortiz de Montellano, P. R. Cytochrome P450: Structure, Mechanism and Biochemistry, 2nd ed., Plenum Press, New York, 1995.
- (b) Dolphin, D.; Traylor, T.G.; Xie, L. Y. Acc. Chem. Res.1997, 30, 251.
- 3. (a) Bonnett, R. Chem. Soc. Rev. 1995, 19.
- (b) Gaud, O.; Granet, R.; Kaouadji, M.; Krausz, P.; Blais, J. C.; Bolbach, G. Can. J. Chem. 1996, 74, 481.
- (c) Driaf, K.; Granet, R.; Krausz, P.; Kaouadji, M.; Thomasson, F.; Chulia, A. J.; Verneuil, B.; Spiro, M.; Blais, J. C.; Bolbach, G. Can. J. Chem. 1996, 74, 1550.
- 4. Arimura, T. J. Synthetic Org. Chem. Japan 1997, 55, 557.
- 5. (a) Lindsey, J. S.; Prathapan, S.; Johnson, J. E.; Wagner, R. W. Tetrahedron 1994, 50, 8941.
- (b) Drain, C. M.; Nifiatis, F.; Vasenko, A.; Batteas, J. D. Angew. Chem. Int. Ed. 1998, 37, 2344.
- (c) Vicente, M. G. H.; Cancilla, M. T.; Lebrilla, C. B.; Smith, K. M. Chem. Commun. 1998, 2355.
- (d) Biemans, H. A. M.; Rowan, A. E.; Verhoeven, A.; Vanoppen, P.; Latterini, L.; Schenning, J.; Meijer, E. W.; Schryver, F. C.; Nolte, R. J. M. J. Am. Chem. Soc. 1998, 120, 11054.
- (e) Osuka, A.; Shimidzu, H. Angew. Chem. Int. Ed. Engl. 1997, 36, 135.
- 6. Kaim, W.; Schwederski, B. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life , John Wiley & Sons, Chichester, 1995.
- 7. Lindsay Smith, J. R. Metalloporphyrins in Catalytic Oxidations, Sheldon, R. A. Ed., Marcel Dekker, New York, 1994.
- 8. (a) Bedioui, F. Coord. Chem. Rev. 1995, 144, 39.
- (b) Mansuy, D.; Battioni, P.; Battioni, J. P. Eur. J. Biochem. 1989, 184, 267.
- 9. (a) Assis, M. D.; Serra, O. A.; Iamamoto, Y. Inorg. Chim. Acta 1991, 187, 109.
- (b) Assis, M. D.; Mello, A. J. B.; Serra, O. A.; Iamamoto, Y. J. Mol. Catal A: Chemical 1995, 97, 41.
- 10. Meng, G. G.; James, B. R.; Skov, K. A. Can. J. Chem 1994, 72, 1894, and references therein.
- 11. Rocha Gonsalves, A. M. d'A.; Varejăo, J. M. T. B.; Pereira, M. M. J. Heterocyclic Chem. 1991, 28, 635.
- 12. (a) Buchler, J. W.; Kunzel, F. M.; Mayer, U.; Nawra, M. Fresenius J. Anal. Chem. 1994, 348, 371.
- (b) Herrmann, O.; Medhi, H.; Corsini, A. Can. J. Chem. 1978, 56, 1084.
- 13. Adler, A. D.; Longo, F. R.; Kampas, F.; Kim, J. J. Inorg. Nucl. Chem. 1970, 32, 2445.
- 14. (a) Rothemund, P. J. Am. Chem. Soc.1936, 58, 625.
- (b) Rothemund, P. J. Am. Chem. Soc. 1939 61, 2912.
- 15. (a) Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827.
- (b) Lindsey, J. S.; Wagner, R. W. J. Org. Chem. 1989, 54, 828.
- 16. Datta-Gupta, N.; Bardos, T. J. J. Heterocyclic Chem. 1966, 3, 495.
- 17. (a) Bigey, P.; Frau, S.; Lomp, C.; Claparols, C.; Bernadou, J.; Meunier, B. Bull. Soc. Chim. Fr.1996, 13, 679.
- (b) Assis, M. D.; Lindsay Smith, J. R. J. Chem. Soc., Perkin Trans.II 1998, 2221.
- 18. Bonar-Law, R. P. J. Org. Chem 1996, 61, 3623.
- 19. (a) Grishima, L. E.; Brykina, G. D.; Shpigum, O. A. J. Anal. Chem. 1995, 50, 826.
- (b) Jensen, J. J. Chromatog. 1963, 10, 236.
- (c) Chu, T. C. and Chu, E. J. J. Chromatog. 1966, 21, 46.
- 20. (a) Wilkelman, J. Cancer Research, 1962, 22, 589.
- (b) J. Wilkeman, J.; Slater, G.; Grossman, J. Cancer Research 1967, 27, 2060.
- 21. Harada, A.; Shiotsuki, K.; Yamaguchi, H.; Kamachi, M. Inorg. Chem 1995, 34, 1070.
- 22. Silverstain, R. M.; Bassier, G. C.; Morril, T. C. Spectrometric Identification of Organic Compounds, John Wiley and Sons, Inc., 3a ed., 1974 .
- 23. Collman, J. P.; Brauman, J. I.; Doxee, K. M.; Halbert, T. R.; Bunnenberg, E.; Linder, R. E.; Lanar, G. N.; Gaudio, J. D.; Spartalian, K. J. Am. Chem. Soc. 1980, 102, 4182.
- 24. Kumar, R. K.; Balasubramanian, S.; Goldberg, I. Inorg. Chem 1998, 37, 541.
- 25. Bellamy, L. J. The Infrared Spectra of Complex Molecules, John Willey and Sons, New York, 1954, ch. 10.
- 26. Thomas, D. W.; Martell, A. B. J. Am. Chem. Soc. 1959, 81, 5111.
- 27. Meng, G. G.; James, B. R.; Skov, K. A. Can. J. Chem 1994, 72, 1894.
- 28. Fuhrlop, J. H.; Kadish, K. M.; Davis, D. G. J. Am. Chem. Soc. 1982, 95, 5140.
- 29. Bard, A. J. Electrochemical Methods, John Willey and Sons, New York,1980.
- 30. Giraudeau, A.; Callot, H. J.; Gross, M. Inorg. Chem. 1979, 18, 201.
- 31. Arao, T.; Maiya, B. G. Polyhedron 1994, 13,1863.
- 32. Messet, M. J. M.; Shokhirev, N. V.; Enemark, P. D.; Jacobson, S. E.; Walker, F. A. Inorg. Chem. 1996, 35, 5188.
- 33. Kadish, K. M.; Morrison, M. M.; Constant, L. A.; Dickens, L.; Davis, D. G. J. Am. Chem. Soc.1976, 98, 8337.
- 34. (a) Bottomley, L. A.; Kadish, K. Inorg. Chem. 1981, 20, 1348.
- (b) Lexa, D.; Momenteau, M.; Mispeltier, J.; Lhoste, J.M., Bioelectrochem. Bioenerg. 1975, 1, 108.
- 35. (a) Davis, D. G. in "The Porphyrins", D. Dolphin Ed., v.V, ch. 4, 19.
- (b) Constant, L. A.; Davis, D. G. Anal. Chem. 1975, 47, 2253.
Publication Dates
-
Publication in this collection
15 Jan 2001 -
Date of issue
Oct 2000
History
-
Accepted
31 Aug 2000 -
Received
31 May 1999