Resumos
Blendas de dois polímeros semi-cristalinos biorreabsorvíveis, o poli(ácido láctico) (PLLA) e a poli(p-dioxanona) (PPD), foram preparadas por evaporação de solvente em diferentes composições. As blendas foram imersas em tubos de ensaio contendo solução tampão fosfato (pH = 7,4) em um banho termostatizado a 37 ± 1 °C e avaliadas por calorimetria diferencial de varredura (DSC), análise termogravimétrica (TGA) e microscopia eletrônica de varredura (MEV). Foi observado através do estudo in vitro que o PLLA apresenta uma taxa de degradação mais lenta que a PPD e que as blendas apresentam taxa de degradação intermediária, mostrando que é possível variar a taxa de degradação das blendas alterando sua composição.
Blendas poliméricas; polímeros biorreabsorvíveis; degradação in vitro; poli(ácido láctico); PLLA; poli(p-dioxanona); PPD
Blends of two crystalline bioreabsorbable polymers, poly(L-lactic acid) (PLLA) and poly(p-dioxanone) (PPD), have been prepared by solvent casting in different compositions. The blends were immersed in tubes containing a phosphate buffer solution (pH = 7.4) in a thermally controlled bath of 37 ± 1°C and studied by differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and scanning electron microscopy (SEM). It was observed by in vitro study that PLLA has a slower degradation rate than PPD and the blends presented an intermediate degradation rate. This shows that it is possible to vary the degradation rate of the blend by changing its composition.
Polymeric blends; bioreabsorbable polymers; in vitro degradation; poly(lactic acid); PLLA; poly(p-dioxanone); PPD
ARTIGO TÉCNICO CIENTÍFICO
Estudo da Degradação In Vitro de Blendas de Poli(p-dioxanona)/Poli(l-Ácido Láctico) (PPD/PLLA) Preparadas por Evaporação de Solvente
Ana P. T. Pezzin, Cecília A. C. Zavaglia, Eliana A. R. Duek
Faculdade de Engenharia Mecânica, UNICAMP
RESUMO: Blendas de dois polímeros semi-cristalinos biorreabsorvíveis, o poli(ácido láctico) (PLLA) e a poli(p-dioxanona) (PPD), foram preparadas por evaporação de solvente em diferentes composições. As blendas foram imersas em tubos de ensaio contendo solução tampão fosfato (pH = 7,4) em um banho termostatizado a 37 ± 1 °C e avaliadas por calorimetria diferencial de varredura (DSC), análise termogravimétrica (TGA) e microscopia eletrônica de varredura (MEV). Foi observado através do estudo in vitro que o PLLA apresenta uma taxa de degradação mais lenta que a PPD e que as blendas apresentam taxa de degradação intermediária, mostrando que é possível variar a taxa de degradação das blendas alterando sua composição.
Palavras-chave: Blendas poliméricas, polímeros biorreabsorvíveis, degradação in vitro, poli(ácido láctico), PLLA, poli(p-dioxanona), PPD.
Degradation of Poly (L-Lactic Acid) (PLLA) and Poly-p-Dioxanone (PPD) Blends Prepared by Casting: In Vitro Study
Abstract: Blends of two crystalline bioreabsorbable polymers, poly(L-lactic acid) (PLLA) and poly(p-dioxanone) (PPD), have been prepared by solvent casting in different compositions. The blends were immersed in tubes containing a phosphate buffer solution (pH = 7.4) in a thermally controlled bath of 37 ± 1°C and studied by differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and scanning electron microscopy (SEM). It was observed by in vitro study that PLLA has a slower degradation rate than PPD and the blends presented an intermediate degradation rate. This shows that it is possible to vary the degradation rate of the blend by changing its composition.
Keywords: Polymeric blends, bioreabsorbable polymers, in vitro degradation, poly(lactic acid), PLLA, poly(p-dioxanone), PPD.
Introdução
O poli(L- ácido láctico) (PLLA) e a poli(para-dioxanona) (PPD) são polímeros da classe dos poli(a-hidróxi ácidos), uma das famílias de polímeros mais atrativas e promissoras, pois além de biorreabsorvíveis, eles também são biocompatíveis, podendo ser utilizados em diversas aplicações na área médica[1]. A presença das ligações éster tornam os poli(a-hidróxi ácidos) hidroliticamente instáveis, podendo ser degradados em contato com os fluidos corpóreos, resultando em produtos que são reabsorvidos pelo organismo fazendo parte do metabolismo de carboidratos[2]. Nos últimos anos, os polímeros biorreabsorvíveis ganharam uma importância crescente na área médica sendo utilizados em um amplo número de aplicações no corpo humano, tais como, suturas cirúrgicas, sistemas para liberação controlada de drogas, peles artificiais, guias para nervos, veias e artérias artificiais e dispositivos ortopédicos[3]. Geralmente, para os polímeros serem utilizados como biomateriais, é necessário satisfazer uma série de requisitos, os quais nem sempre são atendidos por um único polímero. Uma opção para o desenvolvimento de novos materiais é a obtenção de blendas poliméricas, as quais exibem combinações de propriedades muitas vezes superiores às dos componentes puros[4, 5]. Algumas características, como propriedades mecânicas e degradação podem ser modificadas através da escolha favorável do segundo componente. A degradação dos poliésteres alifáticos tanto in vitro, quanto in vivo, foi investigada por muitos autores[6-9]. Sabe-se que a massa molar, a polidispersividade, o grau de cristalinidade, a morfologia, a história térmica e a estrutura química dos polímeros são fatores que influenciam consideravelmente a taxa de degradação[7]. Polímeros com massa molar mais baixa degradam mais rápido que os de massa molar mais alta[8] e os polímeros ramificados degradam muito mais rápido do que os lineares. Contudo, o mecanismo da degradação não está totalmente compreendido. Um estudo mais detalhado sobre a degradação do PLLA mostra que esse fenômeno ocorre de maneira heterogênea, sendo mais rápido nos domínios amorfos do que nos cristalinos e mais rápido no interior do que na superfície, devido a uma auto-catálise ácida[9]. O entendimento da degradação preferencial no interior é baseado no estudo in vitro, mimetizando o meio fisiológico. O polímero é imerso em um meio aquoso, onde ocorre a absorção de água e inicia-se a clivagem hidrolítica das ligações éster, ocasionando uma diminuição da massa molar. No início, a degradação é mais rápida na superfície do que no centro, devido ao gradiente de absorção de água. Os produtos da degradação são formados tanto na superfície quanto no centro, mas aqueles localizados perto da superfície dissolvem-se mais facilmente no meio, pois os localizados no interior devem difundir-se através da massa. Logo, a concentração de grupamentos ácido carboxílicos aumenta muito mais no interior do que na superfície, catalisando a degradação. O objetivo deste trabalho foi o estudo da degradação in vitro de blendas de poli(L- ácido láctico) (PLLA) e poli(para-dioxanona) (PPD) preparadas por evaporação de solvente, com intuito de se obter materiais com propriedades otimizadas para serem utilizados na área médica.
Experimental
Obtenção das blendas
Os polímeros utilizados neste trabalho foram o poli(L-ácido láctico) (PLLA) de massa molar (MW = 100.000 g.mol-1) (Medisorb - Dupont) e a poli(p-dioxanona) (PPD), obtida na forma de sutura violeta (Ethicon, Inc), cujas estruturas químicas encontram-se, respectivamente, na Figura 1. O corante foi previamente extraído em cloreto de metileno com agitação à temperatura ambiente por cerca de 24 horas. Foram preparadas blendas de PPD/PLLA por evaporação de solvente a temperatura ambiente nas composições (0/100), (20/80), (40/60), (50/50), (60/40), (80/20) e (100/0) % (m/m). O PLLA e a PPD foram dissolvidos separadamente em hexafluorisopropanol (HFIP) para formar soluções 10 % (m/v), que foram mantidas sob agitação por 2 horas, até completa homogeneização. As soluções foram misturadas e as misturas foram vertidas em um molde de vidro (50x30x5 mm) e colocadas em uma câmara para evaporação do solvente por 24 horas, com fluxo constante de ar seco. Os filmes foram secos em uma estufa a vácuo a 60 °C durante 24 horas e armazenados em um dessecador, obtendo-se filmes de amostras com a mesma dimensão do molde.

As amostras foram imersas em tubos de ensaios com tampa rosqueada, previamente esterilizados com álcool 70% contendo solução tampão fosfato pH 7,4 a 37 ± 1 °C, sendo retiradas após 1, 2, 4, 6, 8 e 10 semanas. Após tempos determinados, as amostras foram lavadas com água, secas a vácuo e submetidas às técnicas de caracterização citadas a seguir.
Calorimetria diferencial de varredura (DSC)
As curvas de DSC foram realizadas utilizando-se o equipamento STA 409C (NETZSCH). As amostras, pesando em torno de 10 mg, foram aquecidas utilizando o programa: aquecimento de 20 a 200 °C a 10 °C min1; isoterma de 2 min em 200 °C; resfriamento de 200 a 0 °C a 20 °C.min-1; isoterma de 2 min em 0 °C; aquecimento de 0 a 200 °C a 10 °C min1, sob atmosfera de hélio.
Análise termogravimétrica (TGA)
As amostras dos filmes, pesando cerca de 20 mg foram aquecidas de 25 a 450 °C a 10 °C.min-1 sob atmosfera de hélio, utilizando-se o equipamento STA 409C (NETZSCH).
Microscopia eletrônica de varredura (SEM)
Os fragmentos da superfície superior dos filmes e asuperfície de fratura das mesmas (fraturadas em N2 líquido) foram fixados em um suporte metálico e recobertoscom uma fina camada de ouro, utilizando-se um metalizador de amostras BAL-TEC SCD 050. Os fragmentos foram observados ao microscópio eletrônico de varredura JEOL JXA 860A, sob tensão de 10 a 20 kV.
Resultados e Discussão
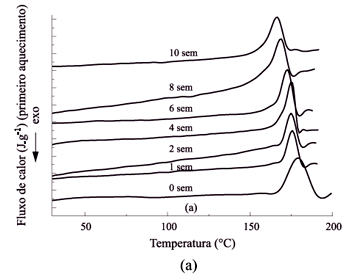
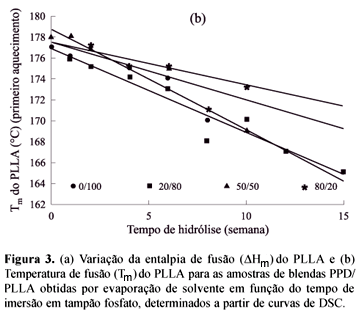
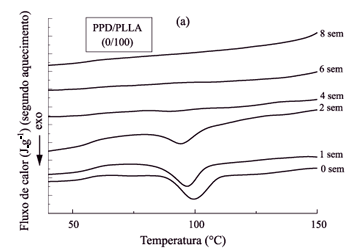
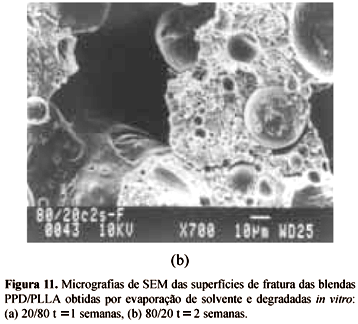
As Figuras 2a e 2b apresentam as curvas de DSC (primeiro aquecimento) para o PLLA e para a PPD, respectivamente, em função do tempo de hidrólise. Na Tabela 1 são apresentados os dados obtidos através das curvas de DSC da Figura 2a para os filmes de PLLA homopolímero. Verifica-se que, após 4 semanas imersos em tampão fosfato, ocorreu um aumento de 43 % na entalpia de fusão (DHm) do PLLA (primeiro aquecimento) e um aumento de mais 25 % neste mesmo valor de 4 para 10 semanas. A Tabela 2 apresenta os dados obtidos através das curvas de DSC da Figura 2b para a PPD homopolímero. O estudo da degradação da PPD mostra um aumento do DHm da PPD de 13 % após 1 semana em tampão fosfato, que se manteve praticamente constante até 4 semanas, indicando que a degradação da PPD é mais rápida no início do processo[10]. A Figura 3a apresenta a entalpia de fusão (DHm) do PLLA para algumas blendas no primeiro aquecimento. Observa-se que o DHm aumenta em função do tempo de imersão em tampão fosfato para as blendas e para os homopolímeros. O aumento da entalpia de fusão significa um aumento na cristalinidade do material, característica desses poliésteres. Até duas semanas em tampão fosfato, verifica-se um aumento mais acentuado do DHm, indicando que a degradação é mais rápida no início do processo. Após esse período, o DHm continua a aumentar, mas não tão significativamente, tendendo a estabilizar. Como as curvas foram normalizadas, é possível perceber que no início do processo, quanto maior a quantidade de PPD, mais acentuado é o aumento do DHm do PLLA. O aumento de DHm indica um aumento no grau de cristalinidade[11,12]. Desta forma, as curvas do DHm do PLLA em função do tempo de hidrólise ilustram a dependência do grau de cristalinidade do PLLA com o tempo de imersão em tampão fosfato. Estes resultados estão de acordo com os dados relatados na literatura com respeito ao aumento da cristalinidade do PLLA[13, 14] e da PPD[12,15]. Lin e colaboradores[12] e Sabino e colaboradores[15] relataram que após a hidrólise em solução tampão fosfato, o grau de cristalinidade da PPD também aumentou. Chu & Browning[16] também observaram aumento de cristalinidade para o PGA, após a degradação hidrolítica. O aumento da cristalinidade do PLLA com o tempo de degradação foi atribuído a cisões hidrolíticas dos segmentos das cadeias e consequente diminuição da massa molar. Tais cisões resultam em um menor enovelamento dos segmentos de cadeia localizados nas regiões amorfas[13,14]. Na Figura 3bpode-se constatar uma diminuição da Tm do PLLA para as blendas em função do tempo de hidrólise, concordando com os resultados da literatura[9,13,14,17]. Por exemplo, na blenda 20/80 a temperatura de fusão do PLLA (primeiro aquecimento) decresce de 177 para 165 °C após 15 semanas em tampão fosfato. Para o PLLA homopolímero a Tm do PLLA diminui de 178 para 169 °C, após 10 semanas em tampão fosfato. Estes resultados podem ser explicados pelo fato de que, com as cisões hidrolíticas dos segmentos das cadeias, ocorre uma recristalização, formando cristais menores, fazendo com que ocorra a fusão dos cristais a uma temperatura mais baixa. Após algumas semanas de hidrólise, observa-se o desaparecimento da temperatura de cristalização (Tc)do PLLA e da PPD, para as blendas 0/100 e 20/80. Na Figura 4a, a Tc do PLLA é verificada até duas semanas, enquanto que na blenda 20/80, a Tc de ambos os polímeros é observada somente até 6 semanas. Com o aumento da degradação, ocorre um rearranjo das cadeias poliméricas, formando novos cristais e resultando no aumento do grau de cristalinidade. Após algumas semanas de degradação, diminui a possibilidade de formar cristais durante o segundo aquecimento, pois o material já se encontra mais cristalino. Para as blendas PPD/PLLA (80/20) há evidências de um segundo pico de fusão para a PPD, o qual se separa do primeiro em função do tempo de degradação. Isto pode ser observado nas curvas de DSC, apresentadas na Figura 4b. Uma explicação para este resultado é que pode estar ocorrendo recristalização dos cristais de PPD durante a corrida do DSC[14]. Pode-se verificar que isto ocorre somente nas blendas com alta concentração de PPD. Pezzin e colaboradores[18] explicaram a ocorrência de dois pontos de fusão para PPD, através da obtenção do fluxo de calor não reversível para uma corrida única (aquecimento) de uma amostra cristalizada a 75 °C por 60 min. Foram observados fortes e múltiplos efeitos exotérmicos nesta curva, comprovando a ocorrência de recristalização. As blendas obtidas por evaporação de solvente foram analisadas por TGA. Os termogramas apresentam um único estágio de perda de massa para os dois homopolímeros. A Tabela 3 ilustra os dados obtidos por TGA para todas as blendas. Comparando-se os valores de Ti da PPD (temperatura em que se inicia o estágio de perda de massa da PPD) em função do tempo de hidrólise, verifica-se um decréscimo nesse valor para a PPD homopolímero, confirmando o processo de degradação. A Figura 5 mostra os termogramas da PPD após 0 e 8 semanas em tampão fosfato, ilustrando esse comportamento.Observa-se que essa degradação é rápida no início do processo, após 2 semanas em tampão fosfato (um decréscimo de 7 %), e após 4 semanas, manteve-se estabilizada, confirmando os resultados obtidos por DSC. Este mesmo comportamento é observado também para os valores de Td da PPD (temperatura em que a derivada primeira da curva de perda de massa é máxima). A análise da Ti do PLLA após 4 e 6 semanas em tampão fosfato, mostra um aumento pouco significativo de 7 °C após 4 semanas que se mantém constante após 6 semanas, o que indica que praticamente não se iniciou a degradação do PLLA homopolímero até 6 semanas em tampão fosfato.Verifica-se que a temperatura onde o PLLA inicia o processo de perda de massa (Ti = 334 °C) é mais alta do que a da PPD (Ti = 258 °C). As blendas 20/80, 50/50 e 80/20 apresentaram dois estágios de perda de massa, sendo o primeiro atribuído à PPD e o segundo atribuído ao PLLA. Ao compararmos a Ti do PLLA para as blendas PPD/PLLA 0/100 e 20/80, verifica-se que a adição de 20 % de PPD à blenda diminui a Ti do PLLA em 12 % após quatro semanas de hidrólise(Figura 6). A diminuição da Ti do PLLA indica que o aumento da concentração de PPD acelera a degradação do PLLA. O acréscimo de 50 % de PPD à blenda acelera a degradação do PLLA em 14 % após 6 semanas em tampão fosfato. Entretanto, com 80 % de PPD na blenda, a variação encontrada após 4 semanas foi a mesma, ou seja, 12 %. As morfologias dos filmes antes e após o processo de degradação foram analisadas por microscopia eletrônica de varredura (MEV). O aparecimento de trincas na superfície da PPD ocorre após 2 semanas de hidrólise, com um aumento significativo após 6 semanas. As Figuras 7a e 7b, mostram, respectivamente, a superfície superior do filme de PPD homopolímero, que apresenta uma superfície irregular composta por glóbulos pontiagudos, antes e após 6 semanas em tampão fosfato. A Figura 7c mostra a superfície da fratura do filme após 6 semanas enquanto a Figura 7d mostra a superfície superior da PPD, após 8 semanas de hidrólise. O aparecimento de trincas ocorre após 2 semanas em tampão fosfato, com um aumento significativo após 6 semanas. Essas trincas podem ser verificadas também na superfície da fratura. Após 8 semanas, a PPD já se encontra completamente degradada. As Figuras 8a e 8b mostram a superfície do filme do PLLA homopolímero após 1 e 10 semanas de hidrólise, na qual não se observam trincas ou rachaduras até este período. Macroscopicamente, a aparência das amostras muda durante o processo de degradação. Após 6 semanas de hidrólise, as amostras tornam-se esbranquiçadas. NIJENJUIS e colaboradores[19] atribuíram este fato à recristalização dos polímeros. Estes resultados comprovam que a PPD degrada muito mais rápido que o PLLA. As blendas em todas as composições foram estudadas in vitro, mas serão discutidos dois casos representativos, que são as blendas PPD/PLLA 80/20 e 20/80. Enquanto a blenda 80/20 é formada por esferas de PLLA dispersas em uma matriz de PPD, o oposto ocorre com a blenda 20/80, que é constituída por esferas de PPD dispersas em uma matriz de PLLA (Figura 9). Ao compararmos as micrografias de superfície inferior das duas composições, após a degradação, observa-se uma diferença significativa quanto ao mecanismo de degradação destas blendas. A Figura 9 mostra que a degradação da blenda 80/20 inicia-se pela matriz, composta de PPD. Após 2 semanas, já é possível observar o aparecimento de algumas trincas (Figura 9a). A Figura 9b mostra a superfície superior da blenda após 4 semanas em tampão fosfato, onde se pode observar que a blenda 80/20 apresenta uma matriz de PPD menos estável composta por esferas de PLLA ancoradas na mesma. Após 6 , 8 e 10 semanas em tampão fosfato, a blenda apresenta-se completamente degradada (Figuras 9d, 9f, 9h). É interessante notar que apesar da matriz estar bastante degradada, as esferas de PLLA apresentam-se praticamente intactas. Por outro lado, o mecanismo de degradação da blenda 20/80 é completamente diferente. Como a matriz é formada por PLLA, que possui uma taxa de degradação mais lenta que o PPD, o processo de degradação desta blenda inicia-se nas esferas de PPD e não na matriz. Após 4 semanas de hidrólise, as esferas começam a se "destacar" da matriz (Figura 9a). Após 6 semanas de hidrólise, não se observa uma variação significativa da degradação (Figura 9c). Com 8 semanas, uma boa parte das esferas de PPD já se "destacaram" (Figura 9e), sendo completamente removidas após 12 semanas, de forma que a superfície da amostra apresenta-se cheia de buracos (Figura 9g). A Figura 10 mostra que o mecanismo de degradação é o mesmo para a blenda de composição 50/50, onde se pode verificar que, após 6 semanas em tampão fosfato, o domínio atribuído à PPD apresenta várias rachaduras, enquanto que o domínio do PLLA ainda permanece intacto. As superfícies de fratura das blendas PPD/PLLA 20/80, após uma semana e 80/20, após 2 semanas (Figura 11), mostram que na parte interna do filme, a degradação ocorre mesmo pelo mecanismo citado acima. Ao compararmos as micrografias das Figuras 9c, 10 e 9d referentes, respectivamente, às blendas PPD/PLLA 20/80, 50/50 e 80/20, após 6 semanas de hidrólise, observa-se que o aumento da concentração de PPD acelera a degradação da mesma.
Conclusões
As técnicas de DSC e TGA permitiram mostrar que a degradação da PPD é mais rápida no início do processo e que a PPD degrada muito mais rápido que o PLLA. A degradação acarreta em um aumento do grau de cristalinidade do material, atribuído às cisões hidrolíticas dos segmentos das cadeias. A análise das micrografias obtidas por SEM mostra que o processo de degradação das blendas inicia-se sempre pela PPD e nos permite concluir que o aumento da concentração de PPD acelera a degradação da mesma. Estes resultados mostram que é possível controlar a taxa de degradação de uma blenda bioreabsorvível variando-se sua composição. Este resultado é interessante, pois as blendas podem ser utilizadas em uma série de aplicações na área médica, dependendo da necessidade. As blendas com maior concentração de PPD são mais flexíveis e degradam mais rápido, podendo ser utilizadas em aplicações onde estas propriedades sejam interessantes, enquanto que as blendas com maior concentração de PLLA são mais rígidas e demoram mais a degradar. Estudos não apresentados aqui mostraram que a blenda PPD/PLLA (80/20) com 3% de trietril citrato de sódio é uma excelente prótese de menisco bioreabsorvível.
Agradecimentos
Gostaríamos de agradecer à Fapesp (processo nº 97/03587-8) pela bolsa concedida.
Recebido: 10/08/01
Aprovado: 05/08/02
Autor para correspondência: Eliana A. R. Duek, Faculdade de Engenharia Mecânica, UNICAMP, Caixa Postal 6122, CEP: 13083-970, Campinas, SP. E-mail: eliduek@fem.unicamp.br
- 1. Iannace, S.; Ambrosio, L.; Huang, S.J.; Nicolais, L. - J. Appl. Polym. Sci., 54, p.1525 (1994).
- 2. Beiser, I.H.; Kanat, I.O. - Journal of the American Podiatric Medical Association, 80, p.72 (1990).
- 3. Langer, R.; Cima, L.G.; Tamada, J.A.; Wintermantel, E.- Biomaterials, 11, p.738 (1990).
- 4. Cha, Y.; Pitt, C.G.- Biomaterials, 11, p. 108 (1990).
- 5. Pearce, R.; Marchessault, R.H.- Polymer, 35, p. 3990 (1994).
- 6. Holland, S.J.; Yasin, M.- J.Controlled Rel., 4, p. 155 (1986).
- 7. Dumitriu, S. "Polymeric Biomaterials", Marcel Dekker, New York (1996).
- 8. Lindhardt R. "Biodegradable Polymers for Controlled Release of Drugs", Springer-Verlag., New York (1988).
- 9. Vert, M.; Li, S.; Garreau, H. - J. Controlled Release, 16, p.15 (1991).
- 10. Assis, P.R.V.; Pezzin, A.P.T.; Zavaglia, C.A.C. and Duek, E.A.R..- "Synthesis and characterization of poly(p- dioxanone) membranes prepared by casting and phase separation", in: Anais do VI Simposio Latino Americano de Polímeros (SLAP VI), IV Congresso Ibero Americano de Polímeros (CIP IV) e IV Simposio Chileno de Química Y Físico Química de Polímeros (CHIPOL IV), 1, p. 201, Viña del Mar, Chile (1998).
- 11. Duek, E.A.R.; Zavaglia, C.A.C.; Belangero, W.D.- Polymer, 40, p.6465 (1999).
- 12. Lin, H.L.; Chu, C.C.; Grubb, D. - J. Biomedical Mater. Res., 27, p.153 (1993).
- 13. Tsuji, H.; Ikada, Y. - Polymer Degradation and Stability, 67, p.179 (2000).
- 14. Tsuji, H.; Ikada, Y. - J. Appl. Polym. Sci., 631, p.855 (1997).
- 15. Sabino, M. A.; González, S.; Márquez, L.; Feijoo, J.L.- Polymer Degradation and Stability, 69, p.209 (2000).
- 16. Chu, C.C.; Browning, A.- J. Biomed. Mater. Res., 22, p.699 (1988).
- 17. Migliaresi, C.; Fambri, L.; Cohn, D.- J. Biomater. Sci., Polym. Ed., 5, p.591 (1994).
- 18. Pezzin, A.P.T.; Alberda van Ekenstein, G.O.R.; Duek, E.A.R.- Polymer, 42, p.8303 (2001).
- 19. Nijennhuis, A.J.; Grijpma, D.W.; Pennings, A.J.- Polymer Bulletin, 26, p.71 (1991).
Datas de Publicação
-
Publicação nesta coleção
25 Fev 2003 -
Data do Fascículo
Out 2002
Histórico
-
Aceito
05 Ago 2002 -
Recebido
10 Ago 2001