Resumos
Nanocompósitos a base de poli(etileno-co-acetato de vinila) (EVA) e óxido de sílica (SiO2), com tamanho nanométrico da ordem de 40 nm, foram preparados via intercalação por solução, empregando clorofórmio como solvente. Os nanocompósitos foram caracterizados principalmente pela espectroscopia de ressonância magnética nuclear (RMN), empregando a análise dos núcleos de carbono-13 (matriz polimérica); silício-29 (nanopartícula) e pela determinação do tempo de relaxação spin-rede do núcleo de hidrogênio no eixo rotatório (T1rH) (matriz polimérica). Pelos dados de RMN foi observado que até cerca de 5% de sílica em massa obteve-se um nanocompósito polimérico com boa dispersão da sílica devido a uma forte interação entre a sílica e matriz de EVA.
Nanocompósitos; RMN; EVA; sílica
Nanocomposites of poly(ethylene-co-vinyl acetate)/silica (SiO2) with dimensions of ca. 40 nm were prepared via solution intercalation employing chloroform as a solvent. They were mainly characterized with nuclear magnetic resonance spectroscopy (NMR) employing carbon-13 (polymeric matrix), silicon-29 (nanoparticles) and through the determination of the proton spin-lattice relaxation time in the rotating frame (T1rH) (polymer matrix). From the NMR results it was inferred that up to 5% of silica in mass a well dispersed nanocomposite was obtained, owing to a strong interaction between silica and the EVA matrix.
EVA; silica; nanocomposites; NMR
Obtenção de nanocompósito de EVA/sílica e caracterização por ressonância magnética nuclear no estado sólido
Preparation of EVA/silica nanocomposites characterized with solid state nuclear magnetic resonance
Adriano A. PassosI; Maria I. B. TavaresII; Roberto C. P. NetoIII; Leonardo A. MoreiraIII; Antonio G. FerreiraIV
ICentro de Tecnologia, IMA/UFRJ. Clariant, Duque de Caxias/RJ
IICentro de Tecnologia, IMA/UFRJ. Laboratório de Ressonância Magnética Nuclear, UFSCar
IIICentro de Tecnologia, IMA/UFRJ
IVCentro de Tecnologia, IMA/UFRJ. Laboratório de Ressonância Magnética Nuclear, UFSCar
Autor para correspondência Autor para correspondência: Maria I. B. Tavares Centro de Tecnologia Instituto de Macromoléculas Professora Eloisa Mano Universidade Federal do Rio de Janeiro Bloco J, Ilha do Fundão, Caixa Postal 68525 CEP: 21945-970, Rio de Janeiro, RJ, Brasil E-mail: mibt@ima.ufrj.br
RESUMO
Nanocompósitos a base de poli(etileno-co-acetato de vinila) (EVA) e óxido de sílica (SiO2), com tamanho nanométrico da ordem de 40 nm, foram preparados via intercalação por solução, empregando clorofórmio como solvente. Os nanocompósitos foram caracterizados principalmente pela espectroscopia de ressonância magnética nuclear (RMN), empregando a análise dos núcleos de carbono-13 (matriz polimérica); silício-29 (nanopartícula) e pela determinação do tempo de relaxação spin-rede do núcleo de hidrogênio no eixo rotatório (T1rH) (matriz polimérica). Pelos dados de RMN foi observado que até cerca de 5% de sílica em massa obteve-se um nanocompósito polimérico com boa dispersão da sílica devido a uma forte interação entre a sílica e matriz de EVA.
Palavras-chave: Nanocompósitos, RMN, EVA, sílica.
ABSTRACT
Nanocomposites of poly(ethylene-co-vinyl acetate)/silica (SiO2) with dimensions of ca. 40 nm were prepared via solution intercalation employing chloroform as a solvent. They were mainly characterized with nuclear magnetic resonance spectroscopy (NMR) employing carbon-13 (polymeric matrix), silicon-29 (nanoparticles) and through the determination of the proton spin-lattice relaxation time in the rotating frame (T1rH) (polymer matrix). From the NMR results it was inferred that up to 5% of silica in mass a well dispersed nanocomposite was obtained, owing to a strong interaction between silica and the EVA matrix.
Keywords: EVA, silica, nanocomposites, NMR.
Introdução
A incorporação de cargas inorgânicas em polímeros origina materiais híbridos, nanocompósitos, com maior resistência mecânica, maior estabilidade térmica ou com propriedades ópticas, magnéticas ou elétricas superiores. As cargas tendo dimensões nanométricas (1-100 nm) apresentam uma área de superfície elevada, promovendo melhor dispersão na matriz polimérica e por isso, uma melhoria das propriedades físicas do compósito, que dependem da homogeneidade do material. A preparação de nanocompósitos de matriz polimérica permite, em muitos casos, encontrar um compromisso entre um baixo custo, devido à utilização de menor quantidade de carga, e um elevado nível de desempenho, que pode resultar da sinergia entre os componentes[1-9].
O desenvolvimento de materiais nanocompósitos ressalta a necessidade de se desenvolver métodos absolutos e inovadores de caracterização destes tipos de materiais, já que o estudo dos mecanismos de formação de híbridos, bem como das interações que ocorrem ao nível da interface inorgânica/orgânica são de grande interesse e de fundamental necessidade, para o entendimento do comportamento desses materiais em nível macroscópicos, visto que a morfologia do nanocompósito é o fator que dita as suas propriedades. É sabido que alterações nas propriedades ópticas, elétricas e/ou mecânicas de um material, à medida que se passa da forma macrocristalina para um material constituído por partículas de dimensões nanométricas, são encontradas serem muitas delas mais profundas, que conotam uma nova aplicação para o polímero[1-9]. Como exemplo pode-se citar partículas compósitas constituídas por nanocristalitos de sulfeto de cádmo (CDs) suportados em sílica, estas foram inseridas em copolímeros de estireno e os nanocompósitos formados apresentaram alterações significativas na microestrutura da matriz polimérica e a dispersão destas cargas nos copolímeros promoveu propriedades ópticas, tornando o material semicondutor.
Sabe-se que as nanopartículas por terem áreas superficiais elevadas, quando são dispersas em matrizes poliméricas promovem alterações nas propriedades da matriz, que são relacionadas com a interação química específica entre as cargas e o polímero. Este tipo de interação pode influenciar na dinâmica molecular da matriz polimérica resultando em alterações significativas nas suas propriedades físicas, nomeadamente no seu comportamento térmico e/ou mecânico[1-6].
Um exemplo de modificação nas propriedades de um polímero foi o estudo de obtenção de material híbrido constituído por nanopartículas de sílica (SiO2) inseridas em poliimida. Pelos resultados gerados foi concluído que a formação de ligações químicas entre as nanopartículas e determinados grupos terminais da poliimida, confinam o espaço acessível às cadeias poliméricas. Segundo esses pesquisadores, o confinamento espacial das cadeias poliméricas influencia a dinâmica molecular do polímero, resultando em alterações na sua estabilidade térmica e temperatura de transição vítrea (Tg), devido à reorganização molecular. Outro estudo mostrou que foram descritas alterações da cristalização de matrizes poliméricas semicristalinas quando em nanocompósitos com sílica. Num material híbrido constituído por uma matriz de poliamida contendo fibras ou esferas de SiO2 com dimensões nanométricas foi registrada a influência da morfologia das nanopartículas na cinética de cristalização da poliamida[10].
Sabe-se que a afinidade química reduzida entre as cargas inorgânicas (natureza hidrofílica) e o polímero (predominantemente hidrofóbico) é um aspecto importante a considerar na preparação de nanocompósitos. A compatibilidade das cargas com a matriz polimérica pode ser melhorada por meio da modificação química superficial das partículas. Para este tratamento normalmente é utilizado um agente de derivatização, que promove a compatibilização química entres os componentes por intermédio de ligações de hidrogênio; interações eletrostáticas ou por ligações covalentes na interface inorgânica/orgânica. Na preparação dos nanocompósitos poliméricos existem duas estratégias principais para estabelecer interações químicas e físicas entre os componentes deste material. Uma das estratégias é o tratamento orgânico da superfície das partículas inorgânicas e a outra consiste na inserção prévia de um monômero hidrofílico na cadeia polimérica. Quando a nanocarga é o SiO2 a estratégia mais simples consiste em reações de substituição na superfície da sílica. As partículas assim tratadas se dispersam em solventes orgânicos e podem ser usadas posteriormente em métodos que envolvam polimerização em suspensão ou em emulsão[11-13].
A caracterização de materiais compósitos e recentemente dos nanocompósitos tem a necessidade de gerar o maior número de informações precisas tanto sobre a estrutura química, com relação à morfologia/propriedade, quanto o comportamento dinâmico molecular, que geram respostas sobre o comportamento mecânico dos materiais. Varias técnicas consideradas convencionais são empregadas em conjunto para se obter uma resposta da formação do nanomaterial. Entretanto outras técnicas denominadas como não convencionais, como a ressonância magnética nuclear (RMN), podem ser empregadas devido a sua potencialidade e ser uma técnica alternativa e efetiva na caracterização de nanocompósitos[14-35].
A espectroscopia de ressonância magnética nuclear é uma ferramenta muito potente no estudo de caracterização de materiais, principalmente no estado sólido. Desde a descoberta do estudo por RMN de materiais no estado sólido, houve um avanço das técnicas para obtenção de espectros no estado sólido com alta resolução em comparação aos obtidos em solução. Assim, o estudo de polímeros e materiais poliméricos pode-se desenvolver rapidamente, por consequência muitas informações a cerca da relação estrutura-propriedade podem ser investigadas com maior precisão. Tendo em vista que a RMN no estado sólido é constituída de várias técnicas, que se diferenciam por diferentes sequências de pulsos e geram respostas distintas permitindo obter dados em diferentes escalas de tempo. Isso faz com que o desenvolvimento de novos métodos analíticos seja cada vez mais crescente e gere um grande impulso para a área da Ciência dos materiais poliméricos[14-22]. Quando o material é insolúvel, como, por exemplo, os nanocompósitos poliméricos o estudo da relação morfologia - propriedade é de grande interesse, pois a busca por respostas com relação à homogeneidade; dispersão de fases e interação entre os componentes é de grande interesse para entender o comportamento do nanomaterial. Logo o estudo por RMN desses materiais deve ser realizado no estado sólido[15-23]. Para o estudo de materiais sólidos, a determinação dos tempos de relaxação spin-rede, com constante de tempo (T1H) e spin-rede no eixo rotatório, com constante de tempo T1rH, especialmente este último propiciam a geração de informação pormenorizada sobre a relação da nanoestrutura (morfologia)/propriedades dos materiais híbridos, o que faz com que o uso da RMN no sólido seja bastante atrativo para o estudo e entendimento do comportamento da nanoestrutura em nanocompósitos poliméricos, visto que os tempos de relaxação geram respostas em nível molecular[15, 23-35]. Os tempos de relaxação envolvem o mecanismo pelo qual ocorre o processo de relaxação de um material após ter sido submetido à radiação eletromagnética. O ambiente químico e as interações existentes e/ou formadas nestes materiais interferem no processo de relaxação. Geralmente quando o material torna-se mais rígido em face de interações fortes, como por exemplo, ligação de hidrogênio, o valor do parâmetro de relaxação T1rH diminui, devido à diminuição da proximidade espacial das cadeias, tornando o material com menor mobilidade molecular.
Rotação Segundo o Ângulo Mágico (MAS)
As análises no estado sólido geram espectros com sinais largos, devido à forte interação dipolar entre os núcleos de 1H e 13C e a anisotropia do deslocamento químico. A técnica básica foi desenvolvida para obtenção de espectros de RMN de alta resolução de sólidos, principalmente pela eliminação da interação dipolar e do efeito da anisotropia do deslocamento químico. Para tanto, deve-se girar a amostra num ângulo de 54,74°, que foi denominado ângulo mágico, constituindo-se assim a técnica básica da RMN no estado sólido[14-18]. A combinação do ângulo mágico com o forte desacoplamento de hidrogênio elimina a interação dipolar (Equação 1).

No estado líquido a interação dipolar é anulada devido aos movimentos Brownianos.
A rotação no ângulo mágico minimiza o efeito da anisotropia do deslocamento químico (Equação 2).

Polarização Cruzada (CP)
A baixa abundância isotópica dos núcleos de spin raros, como por exemplo, o núcleo de 13C, magneticamente ativo, dificulta a sua detecção. Pela RMN no estado sólido a detecção destes tipos de núcleos pode ser obtida através da técnica de polarização cruzada[14-18]. Este método se baseia na transferência de polarização de um núcleo de spin abundante, como o hidrogênio, para um núcleo de spin raro, como o núcleo de 13C. Na polarização cruzada a frequência de pulsos aplicada faz com que a precessão dos núcleos de carbono e hidrogênio tenham frequências idênticas em seus respectivos eixos de rotação a um intervalo de tempo limitado, denominado tempo de polarização cruzada, que obedece a condição de Hartman-Hahn (γC B1C = γHB1H).
Polarização Cruzada e Rotação Segundo o Ângulo Mágico (CP/MAS)
A combinação das técnicas de polarização cruzada (CP), rotação segundo o ângulo mágico (MAS) e forte desacoplamento de alta potência de hidrogênio, tornou possível a obtenção de espectros de RMN de alta resolução para sólidos. Esta técnica permite também o aumento da intensidade do sinal de ressonância dos núcleos de spins raros e a diminuição do intervalo de tempo para a repetição da sequência de pulsos[14, 18-22].
Determinação do tempo de relaxação spin-rede do hidrogênio eixo rotatório
Este parâmetro é determinado pelos decaimentos das intensidades dos núcleos de carbonos-13 resolvidos após a técnica de VTC. Os valores deste parâmetro são relacionados à mobilidade molecular, compatibilidade, homogeneidade e dispersão de partículas.
Assim, este trabalho teve como objetivos a geração de nanomateriais a base de EVA/SiO2, empregando sílica modificada superficialmente com composto orgânico, empregando o método de fusão e a caracterização por RMN no estado sólido, já que esta permite tanto a observação da matriz polimérica, pelas análises dos núcleos de carbono e hidrogênio quanto da nanopartícula, nesse caso o núcleo de silício.
Experimental
Preparação do nanocompósitos por fusão
Os nanocompósitos de EVA/SiO2 foram preparados em extrusora dupla rosca, a 180 °C, a velocidade de rotação de 60 rpm com proporções de 5, 10 e 20% em peso de SiO2.
Análise de RMN em solução
Primeiramente o EVA foi caracterizado por RMN em solução, em um espectrômetro VARIAN MERCURY 300, operando a 75 MHz para o núcleo de carbono-13. A análise foi realizada a temperatura ambiente, empregando clorofórmio como solvente, o intervalo de reciclo para a análise quantitativa foi 20 segundos. O EVA analisado possui 19% em peso de grupos acetatos.
Análise de RMN no estado sólido
As análises de RMN foram realizadas em um espectrômetro de RMN de Varian Unity Plus 400, operando a frequência de 100,4 MHz para o núcleo de carbono-13 (13C) e 79,4 MHz para o núcleo de silício-29 (29Si). As análises foram realizadas a temperatura ambiente da sonda (20 °C), a uma velocidade de rotação de 6 kHz. As análises do núcleo de 13C empregaram as técnicas de: rotação da amostra segundo o ângulo mágico (MAS); rotação da amostra segundo o ângulo mágico e polarização cruzada (CPMAS); experimento de variação do tempo de contato durante a polarização cruzada (VTC) e a determinação do tempo de relaxação spin-rede do hidrogênio no eixo rotatório, que foi determinado a partir do decaimento das intensidades dos núcleos de carbono-13 resolvidos durante o experimento de VTC, pelo aumento no tempo de polarização cruzada. As técnicas empregaram pulso de 90°; intervalo de reciclo de 0,3 segundos para a técnica MAS e 2 segundos para as demais técnicas e o número de acúmulos foi 3200. O núcleo de silício-29 foi investigado empregando a técnica de rotação da amostra segundo o ângulo mágico, com intervalo de reciclo de 10 segundos e número de acúmulo de 10000.
Resultados e Discussão
Análise dos nanocompósitos via matriz polimérica
A matriz polimérica foi investigada pela detecção dos espectros de RMN do núcleo de carbono-13, empregando as técnicas básicas MAS e CPMAS e pelo parâmetro de relaxação spin-rede no eixo rotatório do núcleo de hidrogênio, que foi medido indiretamente pelo decaimento dos núcleos de carbono-13, durante o experimento de variação do tempo de contato.
A técnica de 13CMAS foi realizada, empregando um intervalo de reciclo curto (0,3 segundos), suficiente para a observação apenas da região de maior mobilidade. A Figura 1 mostra os espectros de RMN de 13CMAS do EVA (topo) e do EVA com 5% de nanopartículas de sílica (abaixo). Não foram encontradas diferenças nos espectros das duas amostras, isto porque a região de mobilidade dos materiais é a mesma. Assim, em ambos os espectros foram determinados os sinais referentes à região de maior mobilidade do EVA, que consiste da região amorfa do etileno, com sinal em localizado em 31 ppm e a metila de ramificação em 15 ppm e os sinais referentes ao grupo acetato, como a metila em 21 ppm; os sinais em 26 e 35 ppm referentes aos grupos metileno; o sinal em 74 ppm devido ao grupo CH-O e o sinal em 169 ppm referente ao grupo carbonila.
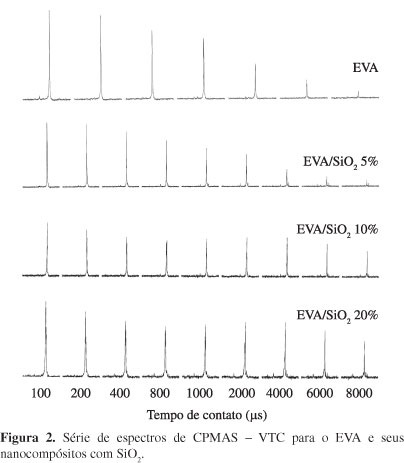
Já que a RMN possui uma variedade de técnicas, o uso de técnicas em conjunto permite gerar informações importantes sobre os materiais. Assim, o experimento de variação do tempo de contato durante a polarização cruzada foi realizado em seguida e este gerou várias informações. A primeira foi em relação à forma de distribuição dos espectros de RMN de 13C CPMAS, com o aumento do tempo de contato, já que os tempos de contato variam de valores curtos (0,1 ms) a valores longos (6 ms). O padrão do filme de EVA e seus nanocompósitos com sílica podem ser observados na Figura 2. Para o nanocompósito com 5% em peso de SiO2, foi observado um decréscimo linear, o que indica uma boa homogeneidade do sistema; enquanto que para 10 e 20% dois domínios foram detectados, mostrando uma não homogeneidade do sistema. A segunda diz respeito à detecção do melhor tempo de contato para a transferência de polarização, gerando o melhor espectro de RMN de 13C CPMAS, que informa sobre todos os tipos de carbono presentes na estrutura e particularmente os mais rígidos. E a terceira informação é a determinação do parâmetro de relaxação spin-rede no eixo rotatório, T1rH. Por este parâmetro pode-se avaliar o comportamento da dinâmica molecular dos nanocompósitos obtidos.
Pelas séries de espectros da Figura 2, o melhor tempo de contato para cada amostra foi o 100 μs para o nano de EVA/SiO25%, mostrando que o material possui um domínio rígido que controla o processo de relaxação, e que este predomina no sistema. Para os sistemas de EVA/SiO210% e EVA/SiO220%, pelo menos dois domínios foram detectados, porque dois tempos de relaxação ótimos foram detectados, um em 100 μs e outro em 1000 ms, evidenciando a formação de dois de mobilidades diferentes, evidenciando que os sistemas possuem heterogeneidade em nível molecular.
Outra informação importante é a determinação do tempo de relaxação spin-rede do hidrogênio no eixo rotatório, T1rH, pelo decaimento das curvas de VTC dos núcleos de carbono-13 resolvidos. A Tabela 1 exibe os valores do tempo de relaxação do hidrogênio no eixo rotatório dos hidrogênios ligados ao o núcleo de carbono-13 localizado em 33 ppm, que é referente proveniente da cadeia polimérica. De acordo com os valores de relaxação determinados para o parâmetro T1rH dos sistemas em estudo, apenas a proporção de 5% em massa de nano sílica gerou um nanocompósito com boa interação entre a sílica e a matriz polimérica, devido à boa dispersão da sílica no polímero, já que foi detectada uma diminuição do valor de relaxação, esta diminuição ocorre quando há interações fortes entre os componentes da mistura. As outras composições mostraram separação de fases e/ou uma não dispersão completa da nanocarga, que ficou clara pela não variação do parâmetro de relaxação, os valores encontrados foram os mesmos para o homopolímero de EVA.
Análise dos nanocompósitos via nanopartícula
O comportamento da nanopartícula foi avaliado pelos espectros de RMN de silício-29 (Si-29) dos nanocompósitos poliméricos, permitindo inferir sobre a influência/interação da nanopartícula com a matriz polimérica. Essa avaliação foi feita em termos de modificação no deslocamento químico do núcleo de Si-29, assim como por meio da mudança na forma dos sinais de Si-29.
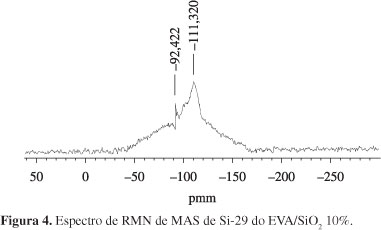
As Figuras 3 e 4 mostram os espectros de RMN de MAS de Si-29 da sílica e do EVA/SiO2 10%.
No espectro da Figura 4 nota-se a perda total da organização do óxido de sílica tanto pela mudança na largura de base do sinal quanto na variação do deslocamento químico do núcleo de silício que variou de -99 para -111 ppm devido à diminuição das ligações com núcleos de oxigênio, mudando sua forma organizacional, indicando que a dispersão deste na cadeia polimérica está aletória, devido às novas interações entre este núcleo e a matriz polimérica, em função da formação de um nanomaterial.
Conclusão
Foi obtido o nanomaterial de EVA/SiO2, apresentando boa dispersão da sílica. Os nanomateriais formados foram caracterizados pelas técnicas de RMN no estado sólido que se mostrou uma ferramenta importante e adequada para a caracterização destes tipos de materiais, pois gera informações ímpares e detalhadas, porque permite analisar o nanocompósitos polimérico tanto pela matriz polimérica quanto pela nanopartícula. O uso do núcleo de silício como sonda de observação da nanocarga mostrou-se efetivo para o acompanhamento das mudanças estruturais ocorridas nos nanomateriais em face tanto da dispersão da nanosílica como pela geração de novas interações entre estas e a matriz polimérica.
Enviado: 22/04/10
Reenviado: 15/07/10
Aceito: 27/08/10
- 1. Kawasumi, M.; Hasegawa, N.; Kato, M.; Usuki, A. & Okada. A. - Macromolecules, 30, p.6333 (1997).
- 2. Li, X. C.; Kang, T.; Cho, W. J.; Lee, J. K. & Há, C. S. - Macrol. Rapid. Commun., 22, p.1306 (2001).
- 3. Nogueira, R. F.; Tavares. M. I. B. & San Gil, R. A. S. - J. Metastable and Crystallinity, 22, p.1210 (2004).
- 4. Nogueira, R. F.; Tavares. M. I. B.; San Gil, R. A. S. & Silva, N. M. - Polym. Test., 24, p.358 (2005).
- 5. Tavares, M. I. B.; Nogueira, R. F.; San Gil, R. A. S.; Preto, M.; Silva, E. O.; Silva, M. B. R. & Miguez, E. - Polym. Test., 26, p.1100 (2007).
- 6. Okamoto, M.; Nam, P. H.; Maiti, P.; Kotada, T.; Nakayama, T.; Takada, M.; Ohshima, M.; Usuki, A.; Hasegawa, N. & Okamoto, H. - Nano Lett., 1, p.503 (2001).
- 7. Gilman, J.; WBourbigot, S.; Shields, J. R.; Nyden, M.; Kashiwagi, T.; Davis, R. D.; Vanderhart, D. L.; Demory, W.; Wilkie, C. A.; Morgan, A. B.; Harris, J. & Lyon, R. E. - J. Mater. Sci., 38, p.4451(2003).
- 8. Gong, F. L.; Zhao, C.; Feng, M.; Qin, H. L. & Yang, M. S. - J. Mater. Sci., 39, p.293 (2004).
- 9. Gong, F. L.; Feng, M.; Zhao, C.; Zhang, S. & Yang, M. - Polym. Test., 23, p.847 (2004).
- 10. Ballard, R. L.; Williams, J. P.; Njus, J. M.; Kiland, B. R. & Soucek, M. D. - Eur. Polym. J., 37, p.381 (2001).
- 11. Hsu, Y. G.; Lin, K. H. & Chiang, I. - Mater. Sci. Eng. B., 87, p.31 (2001).
- 12. Zheng, J.; Ozisik, R. & Siegel, R. W. - Polymer, 46, p.10873 (2005).
- 13. Chang, J. H. & An, Y. U. - J. Polym. Sci., Part B: Polym. Phys., 40, p.326 (2002).
- 14. Zhao, H. & Li, R. K. Y. - Polymer, 47, p.3207 (2006).
- 15. Komoroski, R. A. «High resolution NMR spectroscopy of synthetic polymers in bulk», VCH Publishers, Deerfield Beach (1986).
- 16. McBrierty, V. & Parker, K. J. - "Nuclear magnetic resonance in solid polymers", Cambridge University Press, Great Britain (1993).
- 17. Stejskal, E. O. & Memory, J. D. - "High resolution NMR in the solid state", Oxford University Press, New York (1994).
- 18. Bovey, F. A. & Mirau, P. A. - "NMR of polymers", Academic Press, New York (1996).
- 19. Stael, G. C.; D´Almeida, J. R. M.; Menezes, S. M. C. & Tavares, M. I. B. - Polym. Test., 17, p.147 (1998).
- 20. Harris, R. K. - "NMR studies of solid polymer", in: Polymer spectroscopy, Fawcell, A. H. (ed), John Wiley & Sons, England (1996).
- 21. Tavares, M. I. B. & Monteiro, E. E. C. - Polym. Test., 14, p.273 (1995).
- 22. Silva, N. M. & Tavares, M. I. B. - J. Appl. Polym. Sci., 60, p.663 (1996).
- 23. Stael, G. C.; D´Almeida, J. R. M. & Tavares, M. I. B. - Polym. Test., 19, p.251 (2000).
- 24. Garcia, F. G.; Miguez, E. & Tavares, M. I. B. - Polímeros, 18, p.45 (2008).
- 25. Tavares, M. I. B. - Polym. Test., 19, p.899 (2000).
- 26. Souza, C. M. G.; Tavares, M. I. B. - J. Appl. Polym. Sci., 86, p.116 (2002).
- 27. Dornelas, C. B.; Resende, D. K.; Tavares, M. I. B.; Cabral, L. M. & Gomes, A. S. Polímeros, 18, p.187 (2008).
- 28. Bruno, M.; Tavares, M. I. B.; Motta, L.; Miguez, E.; Preto, M. & Fernandez, A. O. R. - Mater. Res., 11, p.1 (2008).
- 29. Rodrigues, T. C.; Tavares, M. I. B.; Soares, I. L. & Moreira, A. M. - J. Mater., 61, p.34 (2009).
- 30. Silva, E. O.; Tavares, M. I. B. & Nogueira, J. S. - J. Nano Res., 4, p.117 (2009).
- 31. Arantes, T. M.; Leão, K. V.; Tavares, M. I. B.; Ferreira, A. G.; Longo, E.; Camargo, E. R. - Polym. Test., 28, p.490 (2009).
- 32. Hotta, S. & Paul, D. R. - Polymer, 45, p.7639 (2004).
- 33. Alexandre, M. & Dubois, P. - Mater. Sci. Eng., 28, p.1 (2000).
- 34. Tavares, M. I. B.; Nogueira, R. F.; San Gil, R. A. S.; Preto, M.; Silva, E. O.; Silva, M. B. R. & Miguez, E. - Polym. Test., 26, p.1100 (2007).
- 35. Rodrigues, T. C.; Tavares, M. I. B.; Preto, M.; Soares, I. L. & Moreira, A. C. F. - Inter. J. Polym. Mater., 57, p.1119 (2008).
Autor para correspondência:
Datas de Publicação
-
Publicação nesta coleção
13 Maio 2011 -
Data do Fascículo
2011
Histórico
-
Recebido
22 Abr 2010 -
Revisado
15 Jul 2010 -
Aceito
27 Ago 2010