Resumo
The obtention of silica and cyclodextrin hybrid materials was accomplished by refluxing them in xylol using citric acid as a binding agent. The materials were characterized by infrared spectroscopy, X-ray diffraction, thermogravimetric analysis, scanning electron microscopy, and elemental analysis. Evidence for the docking of cyclodextrins α and γ was substantiated based on the variation in band intensity for groups such as ≡Si-OH. Additional docking evidence includes the displacement of some of the bands that are related to cyclodextrin such as the deformation of the C-H axial bond. The α and γ-CDSi materials were characterized as amorphous compounds. The products obtained in the synthesis showed changes in the decomposition temperatures of their isolated constituents, in which the mass of α and γ-CD docked to the silica surface gave the estimated values of 41% and 47%, respectively. The elemental constituents were shown to be consistent and close to their relative theoretical values. Thermogravimetric analysis showed that a reduction in the percentage of the hybrids was proportional to the amount of lost mass. This new material is an improvement over synthesized organosilane materials because the operator and the environment benefit from a less toxic methodology. In addition, the material has several potential applications in complexation systems with cyclodextrin.
cyclodextrin; functionalization; physicochemical characterization
INTRODUÇÃO
A produção de materiais híbridos é um campo de pesquisa que vem apresentando um desenvolvimento considerável nos últimos anos por viabilizar produtos finais mais eficientes e de baixo custo.
Biocerâmicas desenvolvidas a partir de sílicas mesoporosas vêm sendo amplamente estudadas em aplicações como regeneração de tecidos e liberação controlada de drogas.11 Vallet-Regí, M.; Colilla, M.; González, B.; Chem. Soc. Rev. 2011, 40, 596. Sendo a geração de energia uma das grandes preocupações contemporâneas, o desenvolvimento de materiais carbonáceos associados a estruturas inorgânicas para fins de armazenamento e conversão de energia tem crescido bastante.22 Wanga, H.; Dai, H.; Chem. Soc. Rev. 2013, 42, 3088.
A modificação da sílica (Si) permite a obtenção de compostos com maior versatilidade e com propriedades específicas, relacionadas às espécies ligadas à sua superfície.33 Prado, A. G. S.; Miranda, B. S.; Jacintho, G. V. M.; Surf. Sci. 2003, 542, 276. A sílica funcionalizada com quitosana já foi descrita para ser usada na adsorção de metais pesados.44 Repo, E.; Warchol, J. K.; Bhatnagar, A.; Sillanpää, M.; J. Colloid Interface Sci. 2011, 358, 261.Preparações de polímeros híbridos a partir do 3-glicidoxipropiltrimetoxissilano e epóxi vêm sendo investigadas para uso como matriz em revestimentos.55 Davis, S. R.; Brough, A. R.; Atkinson, A.; J. Non-Cryst. Solids 2003, 315, 197. De forma a proporcionar uma estrutura sólida a materiais de vidro transparente, foi desenvolvido um material a partir da combinação de poli-vinilpirrolidona e sílica gel.66 Motoyuki, T.; Chow, T. Y.; Ohnaka, T., Samura, H.; Saegusa, T.; Polym. Bull. 1992, 29, 653.
Entre as diversas aplicações da sílica pode-se destacar a capacidade de troca catiônica,77 Dias, N. L.; Gushikem, Y.; Sep. Sci. Technol. 1997, 32, 535.,88 Francisco, M. S. P.; Gushikem, Y.; J. Mater. Chem. 2002, 12, 2552. a quelação de espécies,33 Prado, A. G. S.; Miranda, B. S.; Jacintho, G. V. M.; Surf. Sci. 2003, 542, 276. a pré-concentração,99 Padilha, P. M.; Gomes, L. A. M.; Padilha, C. C. F.; Moreira, J. C.; Filho, N. L. D.; Anal. Lett. 1997, 32, 1807. seu uso como fase estacionária para cromatografia,1010 Silva, C. R.; Jardim, I. C. S. F.; Airoldi, C.; J. Chromatogr. A 2001, 913, 65. a adsorção de pesticidas,33 Prado, A. G. S.; Miranda, B. S.; Jacintho, G. V. M.; Surf. Sci. 2003, 542, 276. catálise1111 Wilson, K.; Clark, J. H.; Pure Appl. Chem. 2000, 72, 1313. e uso como sensores eletroquímicos.1212 Fujiwara, S. T.; Pessoa, C. A.; Gushikem, Y.; Electrochim. Acta 2003, 48, 3625.,1313 Ribeiro, E. S.; Rosatto, S. S.; Gushikem, Y.; Kubota, L. T.; J. Solid State Electrochem. 2003, 7, 428.
Uma das maneiras mais usadas para modificar a sílica é através das reações com agentes silantes, também chamados de organossilanos. Devido à baixa acidez de Lewis do átomo de silício, a ligação deste átomo com o átomo de carbono tem caráter apolar. Por isso, estes silanos são extremamente eficientes em reações de imobilização e proporcionam a efetiva ligação da parte orgânica do agente com o suporte inorgânico do óxido. Essa ligação só é possível devido à reatividade dos grupos alcóxidos frente aos silanóis presentes na superfície da sílica.1414 Jal, P. K.; Patel, S.; Mishra, B. K.; Talanta 2004, 62, 1005.
As ciclodextrinas (CDs) são oligossacarídeos cíclicos capazes de formarem complexos de inclusão molecular e alterarem as propriedades físico-químicas de outras moléculas, como a solubilidade aquosa e a estabilidade. São, portanto, boas moléculas candidatas a serem imobilizadas na superfície da sílica, proporcionando um ambiente em que moléculas convidadas poderiam ser incluídas nas cavidades das CDs.
A combinação entre a sílica e as ciclodextrinas (CDSi) pode proporcionar a aplicação destes materiais em diversas áreas, desde a indústria farmacêutica,1515 Pasqua, L.; Veltri, L.; Gabriele, B.; Testa, F.; Salerno, G.; J. Porous Mater. 2013, 4, 20. de alimentos,1616 Saikia, M. D.; Colloids Surf., A 2008, 3, 329. agroquímica,1717 Carvalho, L. B.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2012, 74, 375. em cromatografia,1818 Zhang, Z.; Wu, M.; Wu, R.; Gong, J.; Ou, J.; Zou, H.; Anal. Chem. 2011, 9, 83. na remoção de poluentes,1919 Faraji, H.; Husain, S. W.; Helalizadeh, M.; J. Chromatogr. Sci. 2011, 6, 49. em análises biológicas,2020 Banik, A.; Saikia, M. D.; J. Encapsulation Adsorpt. Sci. 2013, 3, 35. dentre outras. A adsorção de corante têxtil em sistema aquoso e a formação de complexo de inclusão com o agroquímico atrazina já foram testadas em nosso grupo, utilizando-se um material híbrido de sílica e β-CD.1515 Pasqua, L.; Veltri, L.; Gabriele, B.; Testa, F.; Salerno, G.; J. Porous Mater. 2013, 4, 20.,1616 Saikia, M. D.; Colloids Surf., A 2008, 3, 329.
A funcionalização da sílica com CD é normalmente realizada com o uso de organossilanos como agentes de ligação, que apresentam alto valor comercial. Nestes trabalhos são também utilizados reagentes como a dimetilformamida (DMF) e sódio metálico, que elevam os riscos de manipulação, além dos longos tempos de síntese que ocasionam maior consumo energético.1515 Pasqua, L.; Veltri, L.; Gabriele, B.; Testa, F.; Salerno, G.; J. Porous Mater. 2013, 4, 20.,1919 Faraji, H.; Husain, S. W.; Helalizadeh, M.; J. Chromatogr. Sci. 2011, 6, 49.
Sendo assim, o presente trabalho propõe a obtenção de um novo material híbrido entre sílica e α e γ-ciclodextrinas, e a posterior caracterização físico-química destes materiais. A utilização de CDs com tamanhos diferentes pode fazer com que moléculas de diferentes dimensões possam interagir com o material, tornando mais versátil a sua aplicação.
PARTE EXPERIMENTAL
Funcionalização da sílica com α e γ-ciclodextrinas
A funcionalização da sílica foi baseada no trabalho do grupo, publicado por Carvalho e colaboradores,2121 Carvalho, L. B.; Carvalho, T. G.; Magriotis, Z. M.; Ramalho, T. C.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 77. que estudaram o ancoramento da β-CD à matriz inorgânica.
Foram utilizadas 1 g de α e γ-CD da Roquette e 1 g de sílica gel (60G) da Sigma-Aldrich. A preparação dos compostos α-CDSi e γ-CDSi foi realizada em refluxo de 6 h utilizando-se, como agente ligante, 1 g de ácido cítrico (99,5% da Vetec), a fim de substituir os organossilanos que têm custo relativamente maior e 50 mL de xilol (Vetec) como solvente.2121 Carvalho, L. B.; Carvalho, T. G.; Magriotis, Z. M.; Ramalho, T. C.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 77.
Posteriormente, o material foi filtrado e lavado exaustivamente, com acetona (Vetec) e água destilada sob vácuo, e seco em estufa, a 65 ºC, até peso seco.
Caracterização dos materiais
Os produtos de síntese foram caracterizados por espectroscopia na região do infravermelho, difratometria de raios-X, termogravimetria e microscopia eletrônica de varredura. A análise elementar também foi escolhida como técnica de caracterização, a fim de se verificar a quantidade percentual dos elementos presentes no material produzido.
Espectroscopia na região do infravermelho
Utilizou-se um espectrofotômetro DigilabExcalibur, série FTS 3000, com transformada de Fourier, para obtenção dos espectros de absorção na região do infravermelho, empregando-se janelas de KBr.
97 mg de KBr e 3 mg do material analisado foram previamente secos em estufa a 60 ºC, por 1 h, e mantidos em dessecador. Posteriormente foram levados a prensa hidráulica, por 10 s, sob uma pressão de 3 ton. Os espectros foram obtidos na faixa de 4000 a 400 cm-1, com resolução de 8 cm-1 e número de acumulações igual a 32 scans.
Difratometria de raios-X
Na difratometria de raios-X (RX) dos produtos e seus precursores utilizou-se uma fonte de Cu-Kα em 2Θ, λ = 1,5406 Å na região compreendida entre 5 e 80º, corrente do tubo de 40 mA, voltagem de 40 kV e varredura de 20 min-1, num difratômetro URD-6 Carl Zeiss, pertencente ao Departamento de Engenharia Química da Universidade Federal de São Carlos.
Análise termogravimétrica
As curvas de termogravimetria (TG) dos materiais foram obtidas sob atmosfera de N2, utilizando-se um aparelho Shimadzu Modelo 60 AH, com temperaturas variando entre 25 e 600 ºC, com fluxo de gás de 30 mL min-1, massa inicial da amostra de cerca de 5 mg e taxa de aquecimento de 10 ºC min-1.
Microscopia eletrônica de varredura
A morfologia do material funcionalizado foi obtida utilizando-se um microscópio eletrônico de varredura LEO EVO 40XVP. As amostras foram fixadas em fita de carbono dupla-face e colocadas em stubs montados com suportes de alumínio. Posteriormente as amostras foram submetidas a banho de ouro coloidal num evaporador Balzers SCD 050, sob atmosfera de argônio.
Análise elementar
A análise elementar foi realizada em um analisador universal da marca Elementar, modelo VarioMicro Cube. Foram utilizados no analisador os gases de arraste e ignição, hélio e oxigênio, respectivamente. Amostras de 2 mg foram acondicionadas em cápsulas de estanho e completamente incineradas a 1200 ºC.
RESULTADOS E DISCUSSÃO
Espectroscopia na região do infravermelho
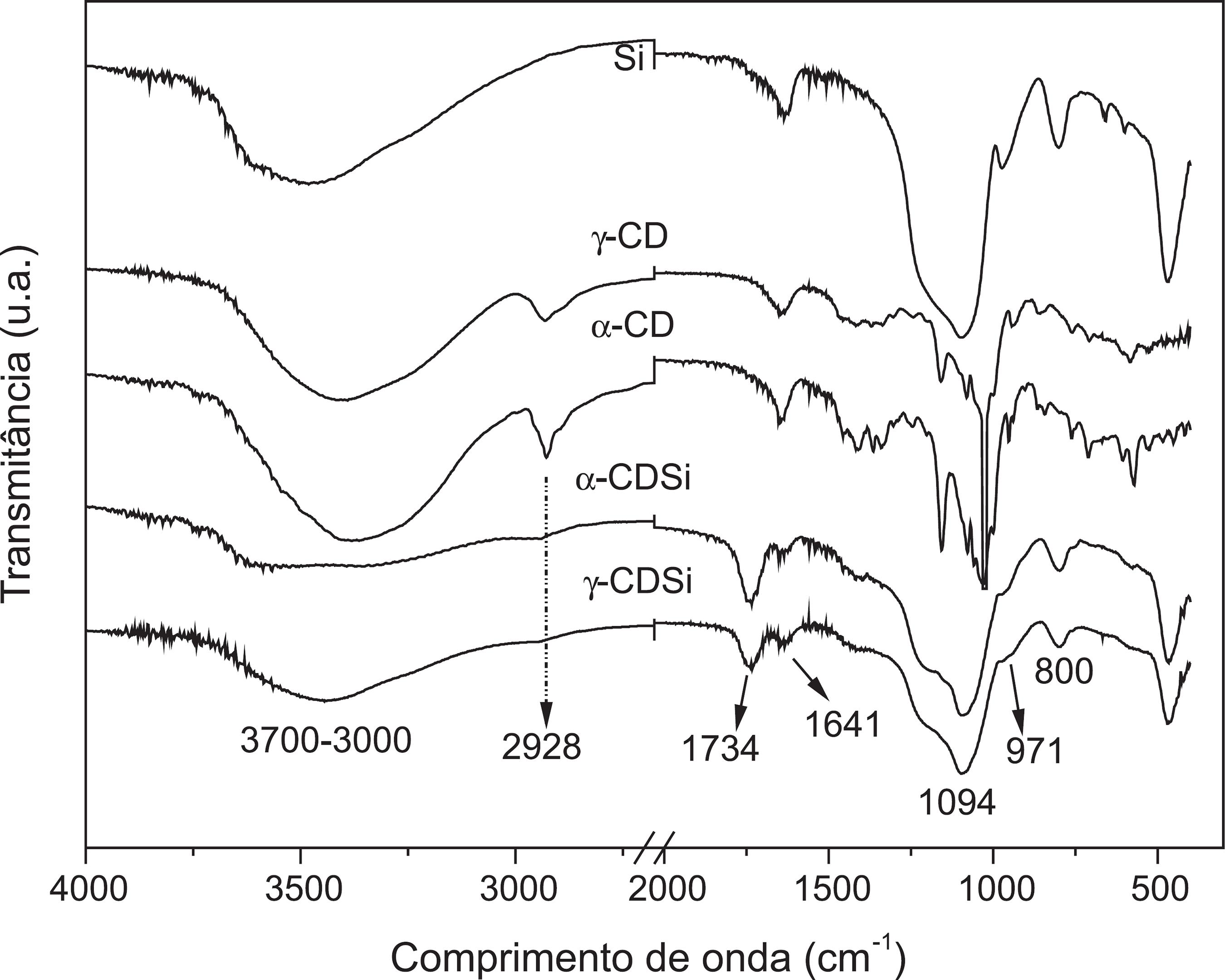
Nas análises feitas por espectroscopia na região do infravermelho (IV), a variação da intensidade de algumas bandas de absorção e alguns deslocamentos em outras fazem com que fiquem evidenciados os suportes da α e da γ-CD à superfície da sílica.
Nos espectros de IV para os precursores e produtos sintetizados (α-CDSi e γ-CDSi), as bandas de absorção em 3700-3000 cm-1 (Figura 1) são características de estiramentos O-H pertencentes aos grupos silanois, à deformação axial das hidroxilas livres e da água remanescente adsorvida.2222 Silverstein, R. M.; Webster, F. X.; Kiemle, D. J.; Spectrometric identification of organic compounds, 7 th ed., Willey: New York, 2005.
A presença da banda em 1734 cm-1 é referente à carbonila, que deve ser dos resíduos de ácido cítrico associados à matriz inorgânica ou a própria CD. Em 1641 cm-1 observa-se uma leve alteração na intensidade das vibrações angulares das moléculas de água,2222 Silverstein, R. M.; Webster, F. X.; Kiemle, D. J.; Spectrometric identification of organic compounds, 7 th ed., Willey: New York, 2005. cujas alterações podem estar relacionadas a algumas interações mais fortes que estas moléculas fazem com a estrutura do composto sintetizado (α-CDSi).
Em 2928 cm-1, a banda de absorção é atribuída à vibração de deformação axial de C-H provenientes de estruturas da α-CD.2222 Silverstein, R. M.; Webster, F. X.; Kiemle, D. J.; Spectrometric identification of organic compounds, 7 th ed., Willey: New York, 2005. Ligeiras alterações de seu deslocamento é um indício da modificação da sílica com a α-CD.
Em 971 cm-1 é observada a banda característica correspondente ao grupamento ≡Si-OH. Com o processo de funcionalização da superfície da sílica, esta banda tem sua intensidade reduzida, pois deve estar ocorrendo uma redução dos grupos silanois livres que cobrem a superfície da sílica. Em 800 cm-1 também ocorre considerável alteração na intensidade da banda característica aos estiramentos realizados por grupos silanois (Si-OH).2222 Silverstein, R. M.; Webster, F. X.; Kiemle, D. J.; Spectrometric identification of organic compounds, 7 th ed., Willey: New York, 2005. Os grupos silanois proporcionam à sílica alta reatividade, pois são conhecidos como centros reativos que possibilitam a imobilização de moléculas.2323 Corriu, R. J. P.; Moreau, J. J. E.; Thepot, P.; Man, M. W. C.; Chem. Mater. 1996, 8, 100.,2424 Dou, B.; Hu, Q.; Li, L.; Qiao, S.; Hao, Z.; J. Hazard. Mater. 2011, 186, 1615. Dessa forma, é provável que os grupos silanois estejam sendo utilizados nas ligações de ancoramento da CD na superfície da sílica.
Difratometria de raios-X
Os resultados da difratometria de raios-X referentes aos precursores e aos materiais sintetizados (Figura 2) mostram mudanças na estrutura cristalina dos materiais, como mencionado por Carvalho e colaboradores para a β-CD.2121 Carvalho, L. B.; Carvalho, T. G.; Magriotis, Z. M.; Ramalho, T. C.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 77. Enquanto os precursores ácido cítrico, α-CD e γ-CD apresentam cristalinidade característica, inerente desses materiais, os compostos α-CDSi e γ-CDSi não fornecem nenhum pico que caracterize essa cristalinidade, indicando que o material produzido é completamente amorfo, assim como a sílica utilizada neste estudo.1717 Carvalho, L. B.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2012, 74, 375.
Difratograma de raios-X obtido para os precursores ácido cítrico (A), γ-CD (B), α-CD (C) e Si (D) e para os materiais sintetizados α-CDSi (E), γ-CDSi (F)
Análise termogravimétrica
A perda de massa do híbrido entre sílica e CD e de seus precursores foi avaliada possibilitando, assim, estimar a massa de CD funcionalizada à superfície da sílica. A massa de CD ancorada foi de aproximadamente 41% da massa total para o primeiro composto (α-CDSi) (Figura 3E) e aproximadamente 47% para o segundo composto sintetizado (γ-CDSi) (Figura 3D). Carvalho e colaboradores2121 Carvalho, L. B.; Carvalho, T. G.; Magriotis, Z. M.; Ramalho, T. C.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 77.encontraram um valor intermediário para o percentual de massa ancorada, 44%, como era de se esperar para a β-CD que é uma ciclodextrina intermediária.
Curvas termogravimétricas para as amostras de ácido cítrico (A), γ-CD (B), α-CD (C), γ-CDSi (D), α-CDSi (E) e Si (F)
A curva TG da sílica (Figura 3F) apresentou ligeira etapa de perda de massa compreendida entre 25 e 102 ºC, correspondente à perda de moléculas de água de hidratação.
No termograma do material α-CDSi (Figura 3E), observou-se três nítidas etapas de perda de massa, sendo a primeira endotérmica, como mostra a DTA (Figura 4) atribuída à desidratação, entre 25 e 112 ºC com perda de 2% da massa. A segunda e a terceira etapas apresentaram-se sobrepostas no gráfico, porém sua diferenciação é possível de ser observada na curva DTA e são atribuídas à decomposição de estruturas orgânicas ancoradas sobre a superfície da sílica: entre 180 e 380 ºC a decomposição de estruturas com ligações menos intensas e entre 380 e 526 ºC a decomposição de espécies com ligações mais intensas ou mais fortemente associadas à superfície da sílica, somando uma perda de massa de 43%.
A curva TG para a α-CD (Figura 3C) apresenta três regiões de perda de massa, sendo a primeira, entre 25 e 109 ºC (decomposição de 6%), referente à perda de moléculas de água de hidratação. A segunda, entre 270 e 360 ºC (exotérmica), referente à decomposição, estimada em 74%, dos grupos orgânicos. E por fim, a terceira etapa, entre 360 e 535 ºC, atribuída à degradação dos resíduos finais, totalizando 20% de perda.
Para o ácido cítrico (Figura 3A) observamos duas etapas de perda de massa. A primeira, correspondente à perda de moléculas de água de hidratação, entre 160 e 260 ºC, correspondente a 88% de sua decomposição, e a segunda, entre 260 e 420 ºC, atribuída à decomposição dos resíduos finais do ácido.
Os resultados sugerem a interação entre a α-CD e a sílica, pois se observou uma alteração na faixa de temperatura na qual ocorreu a decomposição dos constituintes isolados daquele ancorado à superfície da sílica. Sendo assim, pode-se estimar a massa total de α-CD presente no material α-CDSi que foi, como mencionado anteriormente, de 41% da massa total. Foi observado um comportamento semelhante na análise termogravimétrica realizada com a β-CD.2121 Carvalho, L. B.; Carvalho, T. G.; Magriotis, Z. M.; Ramalho, T. C.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 77.
Na curva TG do material γ-CDSi (Figura 3D), observaram-se três nítidas etapas de perda de massa como mostra a DTA (Figura 4), sendo a primeira atribuída à desidratação, entre 25 e 107 ºC, com perda de 4% da massa. A segunda e a terceira apresentaram-se sobrepostas no gráfico e são atribuídas à decomposição de estruturas orgânicas ancoradas sobre a superfície da sílica: entre 185 e 391 ºC a decomposição de estruturas com ligações menos intensas e entre 391 e 522 ºC a decomposição de espécies com ligações mais intensas ou mais fortemente associadas à superfície da sílica, somando uma perda de massa de 51%, demonstrando a funcionalização da superfície.
A curva TG para a γ-CD (Figura 3 B) apresenta duas regiões de perda de massa, sendo a primeira, entre 25 e 115 ºC (decomposição de 9%), referente à perda de moléculas de água de hidratação. A segunda, entre 265 e 374 ºC (exotérmica), refere-se à decomposição, estimada em 76%, dos grupos orgânicos.
Os resultados vistos sugerem a interação entre a γ-CD e a sílica, pois é observada uma alteração na faixa de temperatura na qual ocorreu a decomposição dos constituintes isolados daquele ancorado à superfície da sílica, fornecendo o suporte necessário para estimar a massa total de γ-CD presente no material γ-CDSi que foi, como mencionado anteriormente, de 47% da massa total.
Microscopia eletrônica de varredura
A técnica de microscopia eletrônica de varredura (MEV) foi utilizada para analisar diretamente a estrutura e a distribuição dos cristais formados pelas amostras de α-CDSi e γ-CDSi, assim como seus precursores α-CD, γ-CD, ácido cítrico e sílica.
As micrografias apresentadas na Figura 5 são referentes às estruturas morfológicas dos precursores utilizados para a síntese dos materiais híbridos. Exceto a sílica (Figura 5B), que é amorfa, os demais materiais (ácido cítrico, α e γ-CD) são cristalinos e apresentam cristais, dimensões e contornos típicos de cada um.
Análise por MEV das amostras de ácido cítrico (A), Si (B), α-CD (C), γ-CD (D), α-CDSi (E) e γ-CDSi (F). Aumento de 1000×
Assim, na análise das micrografias para os materiais α e γ-CDSi (Figura 5E e 5F, respectivamente) fica evidente a perda de cristalinidade do material frente aos precursores. A morfologia apresentada pelos híbridos demonstra que o processo de síntese realizado causa uma modificação nas estruturas cristalinas das ciclodextrinas livres (Figura 5C e 5D) e do ligante, ácido cítrico (Figura 5A). As micrografias da α-CDSi e γ-CDSi tiveram perda do padrão característico dos cristais das ciclodextrinas e do agente de ligação ácido cítrico. Houve o aparecimento de um pó amorfo, característico da modificação química superfícial, de dimensões menores que aquelas encontradas nos cristais das substâncias puras. Estes resultados foram semelhantes aos encontrados para o material β-CDSi.2121 Carvalho, L. B.; Carvalho, T. G.; Magriotis, Z. M.; Ramalho, T. C.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 77.
Análise elementar
A composição química pode ser visualizada na Tabela 1, abaixo.
Os valores experimentais que constam na Tabela 1referentes aos precursores e materiais sintetizados mostraram-se coerentes e aproximados em relação aos valores teóricos da análise que são 44% de oxigênio, 49% de carbono e 6% de hidrogênio.
Os resultados obtidos com a análise elementar corroboram com os dados da termogravimetria, pois se tem uma redução percentual nos teores de C e H proporcional à perda de massa encontrada nas curvas TG para a sílica funcionalizada.
CONCLUSÕES
A funcionalização da sílica gel com α e γ ciclodextrinas mostrou-se um processo viável, sendo possível ancorar a CD à superfície da sílica utilizando o ácido cítrico como agente de ligação. Este reagente, além de ambientalmente seguro, apresenta baixo custo e demonstrou ser uma alternativa ao uso de organossilanos mais caros.
Através das referidas caracterizações, confirmou-se o ancoramento das CDs na superfície da sílica a partir de mudanças nos modos vibracionais obtidos nas análises feitas por infravermelho, redução na cristalinidade dos materiais observados por difratometria de raios-X e subsequente mudança nas temperaturas de degradação dos materiais frente aos precursores nas análises termogravimétricas. Também foi possível estimar em 41 e 47% a massa das CDs α e γ, respectivamente, ancoradas na superfície da sílica.
Os novos materiais produzidos, além de demonstrarem relevante estabilidade térmica, possuem potencial para diversas aplicações. Sua aplicação na adsorção de contaminantes aquosos, formação de complexos de inclusão e liberação controlada de agroquímicos já vem sendo testada. Devido à seletividade das CDs na formação de complexos de inclusão, este híbrido pode representar um avanço para os sistemas de liberação de drogas e demais moléculas de interesse comercial, e também no sequestro de moléculas indesejáveis.
AGRADECIMENTOS
Os autores agradecem à UFSCAR e ao CAPQ pelo uso dos equipamentos. À Fapemig (APQ 00687-13) e CAPES (A107/2013) pelas bolsas concedidas e apoio financeiro.
REFERÊNCIAS
-
1Vallet-Regí, M.; Colilla, M.; González, B.; Chem. Soc. Rev 2011, 40, 596.
-
2Wanga, H.; Dai, H.; Chem. Soc. Rev 2013, 42, 3088.
-
3Prado, A. G. S.; Miranda, B. S.; Jacintho, G. V. M.; Surf. Sci. 2003, 542, 276.
-
4Repo, E.; Warchol, J. K.; Bhatnagar, A.; Sillanpää, M.; J. Colloid Interface Sci. 2011, 358, 261.
-
5Davis, S. R.; Brough, A. R.; Atkinson, A.; J. Non-Cryst. Solids 2003, 315, 197.
-
6Motoyuki, T.; Chow, T. Y.; Ohnaka, T., Samura, H.; Saegusa, T.; Polym. Bull. 1992, 29, 653.
-
7Dias, N. L.; Gushikem, Y.; Sep. Sci. Technol. 1997, 32, 535.
-
8Francisco, M. S. P.; Gushikem, Y.; J. Mater. Chem. 2002, 12, 2552.
-
9Padilha, P. M.; Gomes, L. A. M.; Padilha, C. C. F.; Moreira, J. C.; Filho, N. L. D.; Anal. Lett. 1997, 32, 1807.
-
10Silva, C. R.; Jardim, I. C. S. F.; Airoldi, C.; J. Chromatogr. A 2001, 913, 65.
-
11Wilson, K.; Clark, J. H.; Pure Appl. Chem. 2000, 72, 1313.
-
12Fujiwara, S. T.; Pessoa, C. A.; Gushikem, Y.; Electrochim. Acta 2003, 48, 3625.
-
13Ribeiro, E. S.; Rosatto, S. S.; Gushikem, Y.; Kubota, L. T.; J. Solid State Electrochem. 2003, 7, 428.
-
14Jal, P. K.; Patel, S.; Mishra, B. K.; Talanta 2004, 62, 1005.
-
15Pasqua, L.; Veltri, L.; Gabriele, B.; Testa, F.; Salerno, G.; J. Porous Mater 2013, 4, 20.
-
16Saikia, M. D.; Colloids Surf, A 2008, 3, 329.
-
17Carvalho, L. B.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2012, 74, 375.
-
18Zhang, Z.; Wu, M.; Wu, R.; Gong, J.; Ou, J.; Zou, H.; Anal. Chem 2011, 9, 83.
-
19Faraji, H.; Husain, S. W.; Helalizadeh, M.; J. Chromatogr. Sci. 2011, 6, 49.
-
20Banik, A.; Saikia, M. D.; J. Encapsulation Adsorpt. Sci. 2013, 3, 35.
-
21Carvalho, L. B.; Carvalho, T. G.; Magriotis, Z. M.; Ramalho, T. C.; Pinto, L. M. A.; J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 77.
-
22Silverstein, R. M.; Webster, F. X.; Kiemle, D. J.; Spectrometric identification of organic compounds, 7 th ed., Willey: New York, 2005.
-
23Corriu, R. J. P.; Moreau, J. J. E.; Thepot, P.; Man, M. W. C.; Chem. Mater. 1996, 8, 100.
-
24Dou, B.; Hu, Q.; Li, L.; Qiao, S.; Hao, Z.; J. Hazard. Mater. 2011, 186, 1615.
Datas de Publicação
-
Publicação nesta coleção
Set 2015
Histórico
-
Recebido
17 Mar 2015 -
Aceito
06 Maio 2015