Resumo
Depending on formula composition, microemulsions may be used as a vehicle for drug administration. In this work the main applicable parameters used in the development of pharmaceutical microemulsions (ME) are analyzed. The conceptual description of the system, theoretical parameters related to formation of internal phases and some aspects of ME stability are described. The pseudo ternary phase diagram is used to characterize ME boundaries and to describe different structures in several regions of the diagram. Some applications of ME as drug delivery systems for different administration routes are also analyzed. ME offer advantages as drug delivery systems, because they favor drug absorption, being in most cases faster and more efficient than other methods in delivering the same amount of drug.
microemulsions; drug incorporation; drug delivery
microemulsions; drug incorporation; drug delivery
DIVULGAÇÃO
Microemulsões: estrutura e aplicações como sistema de liberação de fármacos
Microemulsions: structure and application as drug delivery systems
Anselmo Gomes de Oliveira* * e-mail: oliveiag@fcfar.unesp.br ; Maria Virgínia Scarpa; Marcos Antonio Correa; Luciane Flávia Rodrigues Cera; Thalita Pedroni Formariz
Departamento de Fármacos e Medicamentos, Faculdade de Ciências Farmacêuticas, Universidade Estadual Paulista "Júlio de Mesquita Filho", CP 502, 14801-902 Araraquara - SP
ABSTRACT
Depending on formula composition, microemulsions may be used as a vehicle for drug administration. In this work the main applicable parameters used in the development of pharmaceutical microemulsions (ME) are analyzed. The conceptual description of the system, theoretical parameters related to formation of internal phases and some aspects of ME stability are described. The pseudo ternary phase diagram is used to characterize ME boundaries and to describe different structures in several regions of the diagram. Some applications of ME as drug delivery systems for different administration routes are also analyzed. ME offer advantages as drug delivery systems, because they favor drug absorption, being in most cases faster and more efficient than other methods in delivering the same amount of drug.
Keywords: microemulsions; drug incorporation; drug delivery.
INTRODUÇÃO
A interação de fármacos com interfaces de agregados supramoleculares de compostos anfifílicos ou de polímeros representa um aspecto importante em tecnologia farmacêutica, pelas implicações biológicas da presença destes compostos em fórmulas farmacêuticas, interferindo no perfil de biodisponibilidade, alterando constantes de ionização de fármacos cujo efeito terapêutico depende substancialmente da fração ionizada da droga ou modificando a estabilidade de grande número de compostos de uso terapêutico1-3.
Independentemente de como se possa definir uma interface, podemos imaginar uma superfície, a partir da qual pode ser descrita uma região externa, contínua, representada pelo meio dispersante, geralmente de natureza aquosa, e outra região, representada pela própria interface, com características particulares e propriedades físico-químicas bastante diferentes das do meio dispersante.
Estas propriedades diferenciadas permitem a utilização destas estruturas em tecnologia farmacêutica, com o objetivo de ampliar as alternativas de aplicação em terapêuticas específicas4. Assim, uma membrana biológica, a superfície de um polímero, micelas, microemulsões e lipossomas representam exemplos de interfaces, em cuja superfície uma série de fenômenos especiais pode ocorrer.
Raramente a experiência clínica com agentes terapêuticos satisfaz as expectativas criadas em torno de resultados pré-clínicos com medicamentos, desde que entre o local de administração e o órgão ou tecido alvo existe uma série de barreiras anatômicas, químicas ou biológicas que contrariam a obtenção do efeito terapêutico desejado. Neste contexto está inserido o conceito dos sistemas transportadores de fármacos, os quais são capazes de compartimentalizar a substância ativa e direcioná-la para os sítios onde deverá exercer o efeito farmacológico, além de poder controlar a velocidade de liberação, sem alterar a estrutura química da molécula transportada4-6. Dessa forma, esses sistemas envolvem as seguintes concepções básicas: a- sistemas matriciais ou monolíticos, nos quais o fármaco está intimamente misturado com os excipientes da fórmula, podendo proporcionar maiores tempos de liberação do mesmo. As micropartículas estão incluídas nesse conceito6,7; b- sistemas reservatórios, nos quais o fármaco encontra-se separado do meio de dissolução através de um revestimento, uma membrana ou uma interface, devendo transpor essa barreira para ser liberado para o meio8-11. As microemulsões são sistemas reservatórios, nos quais a fase interna constitui um microambiente dimensionalmente restrito, com propriedades particulares, podendo ligar ou associar moléculas com diferentes polaridades12,13.
MICROEMULSÕES
As microemulsões (ME) são geralmente caracterizadas como agregados esféricos e com diâmetros menores que 1400 Å, tipicamente da ordem de 100 Å14,15. Apesar da denominação "micro", o sistema envolve gotículas com tamanhos suficientemente pequenos para ser opticamente transparente. Por essa razão, outras denominações, como "submicron emulsion" e "nanoemulsion" têm sido utilizadas para designar esse sistema4. Além dos agregados esféricos, outros tipos de estruturas internas, como as bicontínuas e bicontínuas tubulares foram demonstradas16-21. ME são, de forma geral, definidas como sistemas termodinamicamente estáveis, isotrópicos e transparentes, de dois líquidos imiscíveis, (usualmente água e óleo) estabilizados por um filme de compostos tensoativos, localizados na interface óleo/água3,22-29.
As ME foram introduzidas na literatura por Hoar e Shulman30, em 1943, ao descreverem sistemas transparentes formados espontaneamente quando óleo e água eram misturados com quantidades relativamente grandes de tensoativo iônico misturado a um álcool de cadeia média. Entretanto, o termo microemulsão foi utilizado somente no final da década de 1950 por Schulman e colaboradores31.
Do ponto de vista farmacêutico, as ME podem ser definidas como emulsões transparentes, nas quais um óleo ou um fármaco lipofílico é disperso num meio aquoso (ou vice-versa), contendo um tensoativo, associado ou não a um co-tensoativo apropriado, gerando um sistema termodinamicamente estável (Figura 1).
A formação da microemulsão geralmente envolve a combinação de três a cinco componentes, tais como, tensoativo, fase aquosa, fase oleosa e, quando necessário, o co-tensoativo3,26,29, sendo que a orientação para sistemas O/A ou A/O é dependente das propriedades físico-químicas do tensoativo, traduzidas principalmente pelo seu equilíbrio hidrófilo/lipófilo (EHL)3,22. A principal característica do sistema é formar espontaneamente a fase interna por homogeneização suave dos componentes da fórmula. Sua estabilidade termodinâmica oferece vantagens sobre as dispersões instáveis, tais como as suspensões e emulsões, podendo ser utilizada por tempo muito mais amplo3,24,26.
Considerando que as ME O/A são formadas por gotículas de óleo dispersas em água, contendo o tensoativo na interface óleo/água, elas possuem ótima capacidade de solubilização de substâncias e têm sido usadas para aumentar a estabilidade, a solubilidade e a biodisponibilidade de fármacos, pois permitem a incorporação de vários tipos de compostos na fase interna oleosa (baixa constante dielétrica), na região interfacial (constante dielétrica intermediária entre o óleo e a água) ou na fase externa aquosa (alta constante dielétrica)13,22,23,32.
Teoria da formação
Quando dois líquidos imiscíveis são misturados, mantendo-se agitação constante, as duas fases tendem, inicialmente, a formar gotículas dispersas de um dos líquidos no interior do outro. Quando a agitação cessa, as gotículas tendem a coalescer e os líquidos separam-se novamente. Podemos, então, definir o tempo de vida de uma emulsão, como o tempo decorrido desde o momento em que os líquidos estão completamente homogeneizados, até a separação total do sistema. Assim, o "tempo de vida" é maior quanto melhor for a estabilidade do sistema. Neste ponto interessa-nos analisar mais detalhadamente os motivos que levam à desestabilização da emulsão.
Quando um dos líquidos dividiu-se no interior do outro formou-se uma fase interna, dispersa ou descontínua, rodeada por uma fase externa, dispersante ou contínua. Assim, o processo de emulsificação implica num grande aumento de área interfacial (S1® S2), a qual leva a um aumento brusco da energia livre de superfície (G1® G2)9,27. Este fenômeno, em condições de temperatura constante, pode ser descrito pela Equação:

na qual, gi representa a tensão interfacial entre as fases aquosa e oleosa.
Se considerarmos que o aumento da área interfacial (S1® S2) é imprescindível do ponto de vista tecnológico, uma das alternativas para a estabilização de uma emulsão seria fornecer energia mecânica, continuamente, de modo a manter a área interfacial aumentada8,33. Este fator, embora necessário para a dispersão, por si só não é suficiente, pois vence a barreira da tensão superficial apenas temporariamente, enquanto durar a agitação. Através da Equação 1 pode-se verificar que o caminho mais viável para a estabilização do sistema é a diminuição da tensão interfacial da dispersão, como forma de reduzir a energia livre derivada da expansão da área interfacial. É possível verificar, também, que a estabilidade da emulsão deverá ser maior, quanto menor for a energia livre remanescente da expansão da área interfacial, tendendo a um sistema termodinamicamente estável, caso o aumento da energia livre seja totalmente compensado pela diminuição da tensão interfacial.
Neste aspecto, os agentes tensoativos, com propriedade de diminuir a tensão interfacial entre o óleo e a água, encontram seu papel fundamental na estabilização de emulsões e microemulsões8. Entretanto, esses compostos, com certas exceções, não conseguem diminuir a tensão interfacial a ponto de contrariar totalmente a energia livre de superfície provocada pelo aumento da área interfacial. Dessa forma, as emulsões comuns são consideradas sistemas termodinamicamente instáveis e, no desenvolvimento tecnológico, procura-se utilizar meios que possam retardar pelo maior tempo possível a separação das fases.
Enquanto que as emulsões são estabilizadas por agentes emulsivos comuns (tensoativo), as microemulsões geralmente são adicionadas de um co-tensoativo, cuja função é diminuir a tensão interfacial para valores abaixo dos limites proporcionados pelo emulsivo comum. No entanto, nos casos em que os tensoativos são capazes de cumprir integralmente essa função, a presença dos co-tensoativos não é necessária e a composição da microemulsão restringe-se aos outros três componentes.
As microemulsões diferem das emulsões não somente por serem opticamente transparentes mas, essencialmente, pela estabilidade termodinâmica. Assim, em proporções adequadas entre seus componentes e em condições de temperatura, pressão e força iônica constantes, o sistema forma-se espontaneamente, quando a energia remanescente da interface está próxima de zero (DG ® 0)27.
Além disso, devido às dimensões reduzidas da fase interna, as gotículas possuem altos valores de ângulo de curvatura e de coeficiente de difusão, se comparadas com as emulsões.
Uma das teorias disponíveis para explicar a formação das microemulsões é originada do conceito da "Tensão Negativa Transiente na Interface" 27,34. A energia livre de superfície pode ser definida pela Equação 1 à temperatura, volume e número de moles constantes. Essa equação mostra que DG varia diretamente com a área interfacial e que se, gi for negativo, DG será < 0 e a emulsificação espontânea será possível.
Esse conceito foi utilizado para sistemas microemulsionados de quatro componentes (água, óleo, tensoativo e co-tensoativo) e pode ser analisado a partir de um filme misto monomolecular de tensoativo/co-tensoativo, adsorvido na interface óleo-água. As moléculas orientam-se na interface com as cabeças polares voltadas para a fase aquosa e as cadeias carbônicas para a fase oleosa (Figura 2). Na Figura 2, gO/A representa a tensão interfacial inicial entre o óleo e a água.
Quando aumenta o número de moléculas por unidade de área, elas começam a se comprimirem, umas ao lado das outras, desenvolvendo uma pressão lateral bidimensional (p). A análise do filme mostra que a tensão de superfície (gi) na interface diminui proporcionalmente com o aumento da pressão. Esse fenômeno pode ser expresso pela Equação 2, a qual mostra que quando a repulsão entre as espécies do filme (pressão p) exceder gO/A, gi será negativa:

Desse modo, a variação de energia (gi x DS) torna viável a expansão espontânea da interface. A existência temporária de p > gO/A orienta a força que reduz o tamanho das gotas, a partir de um volume fixo de óleo, até não haver mais necessidade de energia para aumentar a área interfacial. O equilíbrio é atingido quando a tensão negativa volta a zero, em virtude da descompressão das moléculas, com a conseqüente diminuição da pressão na interface.
Uma análise mais criteriosa do sistema mostra que a tensão negativa é resultado não só da alta pressão inicial do filme, mas também da grande diminuição da tensão original entre o óleo e a água (gO/A). Esta depressão começa porque o co-tensoativo (álcool de cadeia média), sendo solúvel tanto na fase oleosa como na interface, vai particionar em ambas as fases, fazendo com que a fração dissolvida no óleo diminua a tensão original de gO/A para (gO/A)a (Figura 3).
A pressão do filme (pG) fica, então, contrariada por uma tensão muito menor (gf), descrita pela Equação 3:

na qual, (gO/A)a e pG, representam a tensão interfacial original entre óleo e água e a pressão no filme antes da curvatura, respectivamente.
Esta seqüência de fatos leva a um mecanismo de curvatura do filme, o qual é aplicável não somente para a formação de microemulsões O/A e A/O, mas também para a magnitude da curvatura do filme.
O filme de tensoativo/co-tensoativo, na interface água-óleo, possui diferentes tensões (ou pressões) em cada lado. Devido a essas diferentes tensões, ocorre a curvatura (Figura 4), dissipando a tensão gradiente até que ambos os lados estejam com tensão equilibrada.
O lado com maior tensão será côncavo e, conseqüentemente, vai englobar o outro líquido deste lado tornando-o a fase interna do sistema. Durante este processo a tensão (gO/A)a, que contraria as pressões nos dois lados do filme monomolecular na interface permanece constante. A pressão total no filme é igual à soma das pressões de cada lado. Assim, pG representa a pressão transiente inicial, resultante da pressão gradiente devido a po e pA através do filme e p é a soma de po e pA (Figura 4).
Diagramas de fase
Quando se modifica progressivamente a composição de uma mistura de solventes miscíveis, as mudanças que se produzirão na solução podem ser traduzidas por importantes variações nas propriedades termodinâmicas dos compostos que a compõe. Por outro lado, as observações dessas propriedades e sua evolução com a composição da solução podem fornecer informações específicas sobre a estrutura do meio.
As microemulsões, com o aspecto de solução homogênea, mas com estrutura micro-heterogênea, constituem um campo de aplicação desse tipo de estudo. Nos sistemas microemulsionados, o objetivo principal é obter uma combinação crítica entre os componentes, de modo a conseguir uma "solubilização" máxima da fase interna ou dispersa33,35,36.
O diagrama de fases descreve em que condição experimental é possível se obter microemulsões e as regiões limites de transição entre emulsões, fases separadas e microemulsões O/A e A/O.
O modo mais usual de descrever esses sistemas de quatro componentes é através do diagrama de fases pseudoternário, onde fase aquosa, fase oleosa e mistura de tensoativo/co-tensoativo são representadas nos vértices do triângulo. Esse tipo de diagrama também suporta outras misturas, como por exemplo óleo, co-tensoativo e água/tensoativo37.
Outra forma de representar o sistema é através do diagrama de fases quaternário, onde tensoativo, co-tensoativo e água estão representados nos vértices do triângulo e o óleo na projeção bidimensional do mesmo36,38. Neste caso, o primeiro triângulo corresponde a zero de óleo e, portanto, a um sistema de micelas mistas contendo água e tensoativo/co-tensoativo. Por isso, muitas vezes as microemulsões são tratadas como uma extensão de soluções micelares.
Se a água e o óleo forem misturados com um tensoativo capaz de produzir uma emulsão leitosa, e essa emulsão for titulada com um co-tensoativo apropriado até formar um sistema opticamente transparente, pode-se determinar a relação tensoativo/co-tensoativo ideal para a obtenção da microemulsão.
O diagrama de fases é definido para cada relação tensoativo/co-tensoativo. Para isso podem-se preparar misturas binárias dos componentes, correspondentes a cada lado do triângulo, como segue:
Mistura emulsiva (T) / fase oleosa (O) em várias proporções, titulando-se com a fase aquosa (A). Essas titulações são representadas, no diagrama, pelas linhas traçadas direcionadas ao infinito de fase aquosa, isto é, convergentes ao vértice do triângulo que representa 100% de fase aquosa (Figura 5a). O mesmo procedimento pode ser utilizado para a mistura emulsiva (T) / fase aquosa (A), a qual é titulada com a fase oleosa (O).
Geralmente esses dois procedimentos são suficientes para se definir o diagrama de fases (Figura 5b). Os pontos da titulação referem-se às modificações verificadas no sistema, tais como, separação de fases, sistema transparente líquido, sistema transparente gel, sistema opaco, etc. Esses pontos são calculados a partir das novas proporções entre os componentes da ME depois da titulação (Figura 5b).
Descrição das regiões do diagrama de fase
A região denominada de ME, na Figura 5b, representa o domínio da existência do sistema opticamente transparente. As regiões 1-5 da Figura 5b descrevem as condições experimentais da existência dos diferentes tipos de sistemas.
Região 1: Existe grande predomínio de fase aquosa. A concentração relativa de tensoativo é pequena <20%. Representativa de ME O/A. Numa diluição infinita com fase aquosa o sistema tende à formação de micelas mistas da mistura emulsiva, contendo a fase oleosa dissolvida em seu interior hidrofóbico. O volume de fase interna é pequeno (Figura 5b). O volume de fase interna é definido pela expressão seguinte3:

na qual w é a relação entre a massa de fase aquosa e a massa total de microemulsão e d é a relação entre as densidades da microemulsão e da fase aquosa.
Região 2: Onde predomina a fase oleosa, é pobre em fase aquosa e em mistura emulsiva. Representativa de ME A/O. Numa diluição infinita tende a formar micelas reversas, compostas por grande parte de fase externa oleosa, com fase aquosa dissolvida nas micelas inversas. O volume de fase interna da ME é pequeno (Figura 5b).
Região 3: Rica em mistura emulsiva, contém muito pouca fase aquosa e fase oleosa (Figura 5b). A estrutura que melhor representa essa região consiste de uma fase contínua onde a mistura de tensoativo/co-tensoativo, óleo e água encontram-se em fase lamelar, na qual o tensoativo e o co-tensoativo organizam-se na interface contínua óleo/água, separando ambas as fases.
Região 4: É intermediária entre as duas regiões (1 e 2) que possuem estruturas bem definidas (Figura 5b). A microestrutura do sistema corresponde a fases bicontínuas, as quais podem explicar a passagem gradual de um sistema O/A para A/O. Esse fenômeno pode ser acompanhado facilmente medindo-se a variação da condutividade, utilizando-se como parâmetro a adição de óleo numa microemulsão A/O ou de água numa microemulsão O/A.
Região 5: Corresponde à região onde o sistema é muito instável e ocorre separação entre as fases aquosa e oleosa.
Para elucidar as modificações microestruturais que acontecem nas várias regiões do diagrama de fases, técnicas de espalhamento de Raios X ("SAXS - Small Angle X-ray Scattering") e de Neutrons à baixo ângulo ("SANS - Small Angle Neutron Scattering"), associadas ou não a técnicas reológicas, têm sido utilizadas em ampla faixa de condições experimentais16,39.
A mistura dos constituintes da ME nem sempre conduzirá a um sistema disperso homogêneo, podendo coexistir diferentes estruturas. Dependendo da natureza e do número de fases líquidas presentes, esses sistemas podem ser classificados no diagrama de fases de acordo com Winsor34,40,41. Região Winsor I, contendo uma fase oleosa em equilíbrio com uma fase emulsionada contendo óleo, água e a maior parte de mistura de tensoativo. Winsor II, também bifásica, constituída de uma fase aquosa em equilíbrio com uma emulsão. Winsor III, trifásica, contendo uma fase aquosa e outra oleosa, separadas por uma fase emulsionada. Winsor IV, correspondente a uma região monofásica, representada por uma emulsão homogênea (Figura 6).
Algumas aplicações de ME como sistema de liberação de fármacos
Substâncias farmacologicamente ativas, quando veiculadas em formas farmacêuticas convencionais, geralmente não conseguem atingir concentração apreciável no tecido alvo do organismo porque, entre o local de aplicação e o local onde deve exercer seu efeito farmacológico, interpõe-se uma série de barreiras biológicas, as quais expõem os tecidos normais do organismo aos efeitos potencialmente tóxicos do fármaco6,24,25. Na atualidade, o estudo de novos sistemas de liberação de fármacos, particularmente as ME, tem sido muito relevante na área farmacêutica, porque pode proporcionar alternativas terapêuticas modernas, farmacologicamente mais eficientes e com efeitos colaterais bastante reduzidos, a partir de fármacos já existentes, muitas vezes pouco utilizados em razão de sua potencial toxicidade12. As ME são adequadas a esses propósitos e têm sido utilizadas, visando a modificação da biodisponibilidade e a diminuição da toxicidade de vários fármacos24,42. De forma geral, a literatura mostra que ME podem proteger fármacos da decomposição ou acelerar esses processos1-3, assim como apresentam grande potencial no uso terapêutico como sistemas reservatórios de fármacos25,26,43 ou são capazes de direcionar fármacos para tecidos ou células específicas do organismo44-48. Trabalhos desenvolvidos em nosso laboratório mostram que ME aumentam significativamente a solubilidade de antiinflamatórios não esteróides25,26,49,50 e antibióticos1-3, funcionando como sistema reservatório que proporciona atividade terapêutica mais intensa e por tempo prolongado. Foi demonstrado que ME constituem um veículo muito mais eficiente que a solução aquosa para a administração de vacinas de DNA por via oral13. ME também têm sido utilizadas na estratégia de aumentar o índice terapêutico dos quimioterápicos antineoplásicos, com a conseqüente redução dos efeitos tóxicos severos desses fármacos45,47.
As propriedades das microemulsões são extremamente variadas, sendo que a grande diversidade de suas aplicações é conseqüência direta desse aspecto. Desde que quantidades apreciáveis de água e óleo podem ser readicionadas às microemulsões, este sistema também constitui um excelente meio de solubilização de compostos polares e apolares, suportando altas concentrações de fármacos nas fases aquosa e oleosa1,51.
A aplicação de microemulsões como sistemas capazes de modular a liberação transdérmica de fármacos e o potencial dessa utilização tem sido descrito e discutido por vários autores52-54.
Microemulsão de liberação tópica
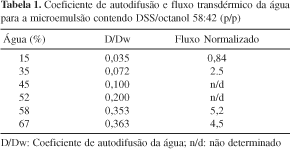
As características e propriedades das ME como veículo de fórmulas farmacêuticas são fundamentais para a liberação tópica de fármacos. Osborne e colaboradores55 caracterizaram um sistema modelo de ME água em óleo com essa finalidade, formado por água/octanol/dodecilsulfosuccinato de sódio (DSS) contendo até 70% de conteúdo de água, tendo verificado que o aumento da proporção de água de 15 a 58% em peso, implica no aumento da média do coeficiente de autodifusão da água (D/Dw) pelo fator de 10 vezes (Tabela 1). D/Dw foi definido como o valor da unidade do coeficiente de autodifusão da água na ME dividido pelo coeficiente de autodifusão da água em água. Dessa forma, a interpretação do valor de D/Dw correspondente a 0,1, na Tabela 1, significa que a água no ambiente de 45% de água na ME difunde com 1/10 da velocidade que a molécula pode difundir no ambiente aquoso puro. Isso implica em que as moléculas de água adicionadas ao sistema estão fortemente ligadas à cabeça polar do DSS e difundem na velocidade de difusão do tensoativo. Entretanto, como os sítios primários de solvatação ao redor do DSS estão saturados, a água livre começará a ser incorporada ao sistema e será viável para difusão e, então, D/Dw irá aumentar de acordo com o aumento do conteúdo de água55. Verifica-se, portanto, que a liberação da porção polar da água da ME é altamente dependente da composição do sistema.
Para o fluxo transdérmico normatizado de água da ME os valores apresentados possuem um perfil semelhante nas mesmas condições de conteúdo de água. Desde que, obviamente, o veículo também afeta a barreira da permeabilidade da pele, quando a quantidade de água aumenta no sistema observa-se aumento de 5 vezes nos valores de fluxo transdérmico para a mesma amostra de pele (Tabela 1)55.
Microemulsão de liberação transdérmica
A pele exerce intensa função de barreira, deixando penetrar apenas algumas substâncias e em determinadas condições. Em razão de seu conteúdo em substâncias tensoativas, as microemulsões podem interagir com o extrato córneo desestruturando a bicamada lipídica do mesmo. Dessa forma, os lipídios passam de uma forma cristalina ordenada para uma forma líquida desordenada, a permeabilidade cutânea é aumentada e a penetração de substâncias, que normalmente não passariam através dessa barreira, fica bastante facilitada.
A permeação in vitro de álcool cetílico, através de pele humana, foi avaliada usando ME A/O. Nesse estudo utilizou-se a célula de Frans e micrótono criostático de micro-seccionamento para quantificar a velocidade e a profundidade da penetração das substâncias, respectivamente56. A penetração do álcool cetílico contido em ME foi cerca de 3,7 vezes maior que num creme e 5 vezes maior que numa loção correspondente, ao longo de 4 h.
Experimentos conduzidos em nosso laboratório mostram que ME O/A, contendo miristato de isopropila como fase interna e mistura de brometo de hexadeciltrimetilamomnio (CTAB)/etanol (1:1) como tensoativo, aumentam significativamente a solubilidade do antiinflamatório não esteróide naproxeno Np, Figura 7.
O espectro de absorção de UV-Vis mostra deslocamento do lmax para regiões de menor comprimento de onda e forte dependência da absorção em relação ao volume de fase interna da microemulsão (Figura 8). Essa diferença espectral reflete o aumento de solubilidade do Np na presença da ME e mostra a dependência da solubilidade em relação ao volume de fase interna do sistema, o que pode interferir diretamente nos parâmetros de liberação in vitro e de atividade antiinflamatória in vivo, já que a disponibilidade do Np vai depender também da velocidade de saída do fármaco da fase interna da ME.
De fato, a liberação in vitro do Np mostra intensa dependência da velocidade de liberação, com decaimento exponencial, em relação ao aumento do volume de fase interna da ME (Figura 9). Esse parâmetro demonstra que é possível controlar a concentração de fármaco disponível no sítio de ação através da modificação do volume de fase interna da ME, o que pode intensificar o efeito antiinflamatório in vivo49.
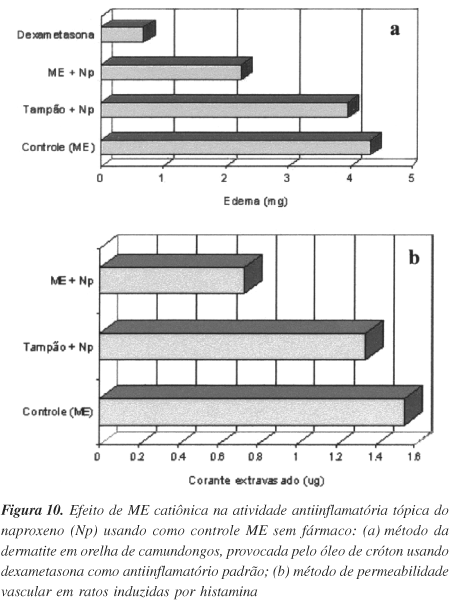
A avaliação da atividade antiinflamatória do Np, em ratos, medindo-se o efeito da aplicação do antiinflamatório incorporado ou não em ME, contra um controle de ME sem fármaco, utilizando-se as metodologias da dermatite por óleo de cróton e a permeabilidade vascular induzida por histamina, revelou que em todos os casos a atividade antiinflamatória foi significativamente mais intensa com ME como veículo, do que a do Np veiculado em sistema tampão (Figura 10)49.
Microemulsão de liberação prolongada
Um sistema de microemulsão biocompatível de liberação prolongada contendo hormônio luteinizante (LH-RH) foi descrito por Gasco e colaboradores52. No estudo, a injeção intramuscular de 500 µg/ml de um análogo de LH-RH veiculado em ME A/O foi aplicada em ratos e o nível de testoterona foi determinado, mostrando um efeito prolongado, com duração entre 10 e 20 dias quando comparado com a administração de 100 µg/kg de LH-RH em solução tamponada.
Foi demonstrado na literatura que a liberação de substâncias ativas dissolvidas na fase interna de ME O/A é mais lenta em relação ao fármaco livre, mostrando a habilidade desse tipo de agregado como sistema reservatório que pode proporcionar efeito prolongado.
Nossos estudos com o antiinflamatório não esteróide piroxicam (Figura 11) utilizando microemulsões catiônicas demonstraram claramente esse fenômeno25,26,50.
A composição das microemulsões utilizadas nos experimentos de liberação in vitro e de avaliação do efeito antiinflamatório in vivo está detalhada na Tabela 2.
O perfil de liberação in vitro do piroxicam em tampão fosfato pH 5,5 (F1), em ME (F2) e em ME contendo polímero carboxivinílico (Carbolpol 940®) (F3), através de membrana de acetato de celulose a 37 ºC mostra que a incorporação do piroxicam na ME catiônica interfere muito significativamente no processo de liberação in vitro, produzindo inibição significativa da velocidade de liberação (Figura 12)25.
A característica mais importante evidenciada nos resultados foi o parâmetro de retenção da substância ativa pela fase interna oleosa da ME, a qual proporciona a liberação constante e regular numa faixa mais ampla de tempo, quando comparada ao controle (Figura 12). O fator de retenção da substância ativa provocado pela ME foi da ordem de 10 vezes. A presença do polímero carboxivinílico acentua o grau de retenção do piroxicam em razão do aumento da viscosidade do sistema, o que dificulta ainda mais a saída do antiinflamatório da fase interna da ME. Esses resultados são importantes, pois fornecem uma base comparativa complementar para a interpretação dos resultados da atividade antiinflamatória in vivo.
A propriedade das ME como sistemas que proporcionam efeito prolongado foi testada in vivo utilizando o método de formação de tecido granulomatoso, ideal para avaliação de processos inflamatórios crônicos (Figura 13). A duração do efeito antiinflamatório foi avaliada utilizando-se piroxicam veiculado em sistema tampão e nos sistemas contendo ME25. Administrou-se dose única de piroxicam (10 mg/kg) por via subcutânea, determinando-se o efeito antiinflamatório depois de 3, 6 e 9 dias. Ao final do terceiro dia observou-se que todas as fórmulas exibiram efeito antiinflamatório significativamente maior do que o grupo controle (p < 0,05), sendo que F2 e F3 mostraram efeito superior ao de F1. Ao final do sexto dia F2 e F3 mantinham o efeito antiinflamatório superior a F1. Os resultados da Figura 13 indicam que a atividade antiinflamatória de F2 e F3 aumentou de 43 e 33%, para 59 e 58%, respectivamente. Por outro lado, F1, contendo piroxicam no sistema tampão, já no terceiro dia exibia atividade máxima de 21,80%, decaindo no sexto dia para 16,80%. Esse fenômeno foi bem evidenciado no final de 9 dias, quando a atividade antiinflamatória provocada por F1 não foi significativa em relação ao controle (p > 0,01). Ficou evidente, também, que a presença do polímero carboxivinílico, embora tenha exercido um efeito adicional na retenção da substância ativa no experimento de liberação in vitro, não mostrou o mesmo perfil na avaliação do efeito antiinflamatório in vivo. Esses resultados mostram claramente o efeito reservatório in vivo proporcionado pela fase interna da microemulsão, o qual demonstra o efeito prolongado da ME como veículo para a administração de antiinflamatórios não esteróides (AINE).
Microemulsão para liberação oral de fármacos
A liberação de peptídeos e proteínas, através de administração oral, possui muitos obstáculos em sua aplicação devido às barreiras enzimáticas e físicas do trato gastrintestinal. Degradação de peptídeos ocorre freqüentemente em vários locais, tais como lume intestinal e mesmo intracelularmente. O direcionamento de certos peptídeos por via linfática pode ser interessante, pois a corrente sanguínea é muito mais eficiente em remover compostos absorvidos do que a linfa54. Teoricamente, os fármacos lipofílicos e com boa solubilidade em triglicerídeos seriam preferencialmente absorvidos pela via linfática.
Estudos mais recentes têm mostrado o grande potencial das ME lipídicas na liberação de fármacos, desde que podem se comportar como sistemas líquidos ou gel, mantendo sua estabilidade termodinâmica12. Esses sistemas são capazes de proteger e favorecer a absorção de fármacos peptídicos via oral. ME lipídicas para liberação de DNA em vacina gênica por via oral, contra a tuberculose, foram desenvolvidas utilizando-se ME A/O contendo derivados de ácidos graxos de cadeia média Captex 200® (dicaprilato/dicaprato de propilenoglicol) e Capmul MCM® (Caprilato/caprato de glicerila) como fase oleosa e mistura de Cremophor EL® (Triricinooleato de polioxietilenoglicerol 35)/fosfatidilcolina de soja como tensoativo13. Plasmídeos contendo gene repórter b-gal ou gene hsp65 ("heat shock protein" de 65kDa, de Mycobacterium leprae) foram incorporados na fase interna da ME. A imunização de camundongos Balb/c com hsp65 mostrou produção de anticorpos IgG total e IgG2a aumentadas em relação aos grupos controle, resposta proliferativa e produção de INF-g específica nas culturas de células esplênicas dos camundongos. Os dados são indicativos que a transfecção foi alcançada e que a resposta imune desenvolvida seja específica e de padrão Th1, adequado para proteção contra a tuberculose13.
Medicamentos desenvolvidos no campo das ME têm permitido a ampliação das possibilidades terapêuticas de fármacos potencialmente tóxicos, como a doxorrubicina57 e ciclosporina58. A ciclosporina, um peptídeo cíclico com 11 aminoácidos, com forte efeito imunossupressor, foi formulada como uma microemulsão completa, pré-concentrada, na qual o tensoativo tem a função de reduzir a variação da absorção da ciclosporina, resultante das modificações do fluxo de bile. Em presença de água a ME forma um sistema homogêneo, opticamente transparente, cujas gotículas da fase interna são menores do que 100 nm, causando um aumento significativo na absorção da ciclosporina58.
CONCLUSÕES
Ao lado das aplicações mencionadas, certamente existem muitas outras aplicáveis na área farmacêutica. Por exemplo, as ME contendo óleos fluorocarbonados em água podem ser usadas como componentes de sangue artificial, já que são capazes de estocar oxigênio e liberá-lo na presença de dióxido de carbono. Desde que os diâmetros das gotículas da fase interna são menores que 1000 Å, isto faz o sistema aplicável em razão de que podem passar livremente através dos capilares. Existem certas vantagens no uso das microemulsões como sistemas de liberação de fármacos, incluindo o fato de que os fármacos são imediatamente absorvidos e, na maioria dos casos, de forma mais rápida e eficiente do que a mesma quantidade de fármaco administrado em comprimidos ou em cápsulas.
REFERÊNCIAS
1. Oliveira, A. G.; Tese de Livre-docência, Universidade Estadual Paulista Júlio de Mesquita Filho, Brasil, 1997.
2. Oliveira, A. G.; Chaimovich, H.; J. Pharm. Pharmacol. 1992, 45, 850.
3. Oliveira, A. G.; Scarpa, M. V.; Chaimovich, H.; J. Pharm. Sci. 1997, 86, 616.
4. Benita, S., ed.; Submicron emulsion in drug targeting and delivery, Harwood Academic: Amsterdam, 1998.
5. Oliveira, A. G.; Scarpa, M. V.; Infarma 2001, 13, 14.
6. Oliveira, A. G.; Scarpa, M. V.; Bueno, J. H. F.; Evangelista, R. C.; Rev. Ciênc. Farm. 1992, 14, 37.
7. Zaniboni, H.; Peres, P. G. P.; Oliveira, A. G.; Rev. Ciênc. Farm. 2001, 22, 263.
8. Attwood, D.; Florence, A. T.; Surfactants Systems: Their Chemistry, Pharmacy and Biology, Chapmam and Hall: London, 1983.
9. Friberg, S. E.; Venable, R. L. Em Encyclopedia of emulsions technology; Becker, P.; ed.; Marcel Dekker: New York, 1983.
10. Lima, E. M.; Lopes, L. B.; Scarpa, M. V.; Oliveira, A. G.; Rev. Ciênc. Farm. 2001, 22, 281.
11. Lima, E. M.; Oliveira, A. G.; Drug. Dev. Ind. Pharm. 2002, 6, 673.
12. Cera, L. F. R.; Dissertação de Mestrado, Universidade Estadual Paulista Júlio de Mesquita Filho, Brasil, 2002.
13. Wargaftig, T. N.; Dissertação de Mestrado, Universidade Estadual Paulista Júlio de Mesquita Filho, Brasil, 2001.
14. Bhargava, H. N.; Narurkar, A.; Lieb, L. M.; Pharm. Technol. 1987, 11, 46.
15. Langevin, D.; Acc.Chem.Res. 1988, 21, 255.
16. Berni, M. G.; Lawrence, C. J.; Machin, D.; Adv. Colloid Interface Sci. 2002, 98, 217.
17. Chen, S. J.; Evans, D. F.; Ninham, B.; J. Phys. Chem. 1984, 88, 1631.
18. Chen, S. J.; Evans, D. F.; Ninham, B; Mitchell, D. J.; Blum, F. D.; Pickup, S.; J. Phys. Chem. 1986, 90, 842.
19. Friberg, S. E.; J. Soc. Cosmet. Chem. 1990, 41, 155.
20. Scriven, L. E. Em Micellization, solubilization and microemulsions; Mittal, K. L., ed.; Plenum Press: New York, 1977, p. 877.
21. Zemb, T. N.; Hyde, S. T.; Derian, P. J.; Barnes, I.; Ninham, B. W.; J. Phys. Chem. 1987, 91, 3814.
22. Constantinides, P. P.; Scarlat, J. P.; Lancaster, C.; Marcello, J.; Marks, G.; Ellens, H.; Smith, P. L.; Pharm. Res. 1994, 11, 1385.
23. Constantinides, P. P.; Welzel, G.; Ellens, H.; Smith, P. L.; Sturgis, S.; Yiv, S. H.; Owen, A. B.; Pharm. Res. 1996, 13, 210.
24. Constantinides, P. P.; Pharm. Res. 1995, 12, 1561.
25. Dalmora, M. E.; Dalmora, S. L.; Oliveira, A. G.; Int. J. Pharm. 2001, 222, 45.
26. Dalmora, M. E.; Oliveira, A. G.; Int. J. Pharm. 1999, 184, 157.
27. Friberg, S. E.; Bothorel, P.; Microemulsions: Structure and dinamics, CRC: Boca Raton, 1988.
28. Ho, H.; Hsiao, C. C.; Sheu, M. T.; J. Pharm. Sci. 1996, 85, 138.
29. Lawrence, M. J.; Eur. J. Drug Metab. Pharmacokinet. 1994, 3, 257.
30. Hoar, T. P.; Schulman, J. H.; Nature 1943, 152, 102.
31. Schulman, J. H.; Stoeckenius, W.; Prince, L. M.; J. Phys. Chem. 1959, 63, 1677.
32. Actis, G. C.; Vopes, R.; Rizzetto, M.; Eur. J. Gastroenterol. Hepatol. 1999, 8, 905.
33. Shah, D. O., ed.; Micelles, microemulsions, and monolayers: Science and technology; Marcel Dekker: New York, 1998.
34. Berthod, A.; J. Chim. Phys. 1983, 80, 407.
35. Garti, N., ed.; Thermal behavior of dispersed systems; Marcel Dekker: New York, 2001.
36. Thevenin, M. A.; Grossiord, M. C.; Poelman, M. C.; Int. J. Pharm. 1996, 137, 177.
37. Khan, A.; Curr. Opin. Colloid Interface Sci. 1996, 5, 614.
38. Rosoff, M. Em Pharmaceutical dosage forms: Disperse systems; Lieberman, H. A.; Martin, M. R.; Banker, G. S., eds.; Marcel Dekker: New York, 1998, vol. 3.
39. Hellweg, T.; Curr. Opin. Colloid Interface Sci. 2002, 7, 50; Hiragi, Y.; Sano, Y.; Matsumoto, T.; J. Synchroton Radiat. 2003, 1, 193; Kang, S. Y.; Seong, B. S.; Jung, H. T.; Biomacromolecules 2003, 4, 360; Steyler, D. C.; Robinson, B. H.; Eastoe, J.; MacDonald, I. Em Micelles, microemulsions, and monolayers: Science and technology; Shah, D. O., ed.; Marcel Dekker: New York, 1998.
40. Binks, B. P.; Cho, W-G.; Fletcher, P. D. I.; Tetsev, D. N.; Langmuir 2000, 16, 1025.
41. Jacovwics, J.; Berthiaume, M. D.; Cosmetics & Toiletries 1993, 108, 65.
42. Constantinides, P. P.; Lancaster, C. M.; Marcello, J.; Chiossome, D. C.; Orner, D.; Hidalgo, I.; Smith, P. L.; Sarkahian, A. B.; Yiv, S. H.; Owen, A. J.; J. Controlled Release 1995, 34, 109.
43. Swenson, E. C.; Curatolo, W. J.; Adv. Drug Delivery Rev. 1992, 8, 39.
44. Maranhão, R. C.; Garicochea, B.; Silva, E. L.; Llacer-Dorlhiac, P.; Cadena, S. M. S.; Coelho, I. J. C.; Meneghetti, J. C.; Pileggi, F. J. C.; Chamone, D.; Cancer Res. 1994, 54, 4660.
45. Maranhão, R. C.; Graziani, S. R.; Brandizzi, L. L.; Coelho, I. J.; Oliveira, M. A.; Hegg, R.; Pinotti, J. A.; Pilleggi, F.; Meneguetti, C. A.; Can. J. Infect. Dis. 1995, 6, 227.
46. Maranhão, R. C.; Hungria, V. T. M.; Chamone, D. A. F.; Chianote, C. S.; Int. J. Hematol. 1996, 64, 127.
47. Maranhão, R. C.; Roland, Y.; Ramiures, J.; Gonçalves, R. P.; Mesquita, C. H.; Pileggi, F.; Lipids 1997, 32, 627.
48. Owens, D. M.; Baillie, G.; Halbert, W. G.; Int. J. Pharm. 2001, 228, 109.
49. Correa, M. A.; Tese de Doutorado, Universidade de São Paulo, Brasil, 1997.
50. Dalmora, M. E.; Dissertação de Mestrado, Universidade Federal de Santa Maria, Brasil, 1997.
51. Takeo, M.; Disperse systems, Willey-VCH: New York, 1999.
52. Gasco, M. R.; Pattarino, F.; Lattanzi, F.; Int. J. Pharm. 1990, 62, 119.
53. Paul, B. K.; Moulik, S. P.; J. Dispersion Sci. Technol. 1997, 18, 301.
54. Sarciaux, J. M.; Acar, L.; Sado, P. A.; Int. J. Pharm. 1995, 120, 127.
55. Osborne, D. W.; Ward, A. J. I.; O' Neill, K. J.; Drug Dev. Ind. Pharm. 1988, 14, 1202.
56. Linn, E. E.; Pohland, R. C.; Byrd, T. K.; Drug Dev. Ind. Pharm. 1990, 16, 899.
57. Mitra, S.; Gaur, U.; Ghosh, P. C.; Maitra, N. A.; J. Controlled Release 2001, 74, 317.
58. http://home.eznet.net/~webtent/drugmonitor.html, acessada em Novembro 2003.
Recebido em 25/9/02; aceito em 27/6/03
- 1. Oliveira, A. G.; Tese de Livre-docência, Universidade Estadual Paulista Júlio de Mesquita Filho, Brasil, 1997.
- 2. Oliveira, A. G.; Chaimovich, H.; J. Pharm. Pharmacol. 1992, 45, 850.
- 3. Oliveira, A. G.; Scarpa, M. V.; Chaimovich, H.; J. Pharm. Sci. 1997, 86, 616.
- 4. Benita, S., ed.; Submicron emulsion in drug targeting and delivery, Harwood Academic: Amsterdam, 1998.
- 5. Oliveira, A. G.; Scarpa, M. V.; Infarma 2001, 13, 14.
- 6. Oliveira, A. G.; Scarpa, M. V.; Bueno, J. H. F.; Evangelista, R. C.; Rev. Ciênc. Farm. 1992, 14, 37.
- 7. Zaniboni, H.; Peres, P. G. P.; Oliveira, A. G.; Rev. Ciênc. Farm. 2001, 22, 263.
- 8. Attwood, D.; Florence, A. T.; Surfactants Systems: Their Chemistry, Pharmacy and Biology, Chapmam and Hall: London, 1983.
- 9. Friberg, S. E.; Venable, R. L. Em Encyclopedia of emulsions technology; Becker, P.; ed.; Marcel Dekker: New York, 1983.
- 10. Lima, E. M.; Lopes, L. B.; Scarpa, M. V.; Oliveira, A. G.; Rev. Ciênc. Farm. 2001, 22, 281.
- 11. Lima, E. M.; Oliveira, A. G.; Drug. Dev. Ind. Pharm 2002, 6, 673.
- 12. Cera, L. F. R.; Dissertação de Mestrado, Universidade Estadual Paulista Júlio de Mesquita Filho, Brasil, 2002.
- 13. Wargaftig, T. N.; Dissertação de Mestrado, Universidade Estadual Paulista Júlio de Mesquita Filho, Brasil, 2001.
- 14. Bhargava, H. N.; Narurkar, A.; Lieb, L. M.; Pharm. Technol. 1987, 11, 46.
- 15. Langevin, D.; Acc.Chem.Res. 1988, 21, 255.
- 16. Berni, M. G.; Lawrence, C. J.; Machin, D.; Adv. Colloid Interface Sci 2002, 98, 217.
- 17. Chen, S. J.; Evans, D. F.; Ninham, B.; J. Phys. Chem. 1984, 88, 1631.
- 18. Chen, S. J.; Evans, D. F.; Ninham, B; Mitchell, D. J.; Blum, F. D.; Pickup, S.; J. Phys. Chem. 1986, 90, 842.
- 19. Friberg, S. E.; J. Soc. Cosmet. Chem. 1990, 41, 155.
- 20. Scriven, L. E. Em Micellization, solubilization and microemulsions; Mittal, K. L., ed.; Plenum Press: New York, 1977, p. 877.
- 21. Zemb, T. N.; Hyde, S. T.; Derian, P. J.; Barnes, I.; Ninham, B. W.; J. Phys. Chem. 1987, 91, 3814.
- 22. Constantinides, P. P.; Scarlat, J. P.; Lancaster, C.; Marcello, J.; Marks, G.; Ellens, H.; Smith, P. L.; Pharm. Res. 1994, 11, 1385.
- 23. Constantinides, P. P.; Welzel, G.; Ellens, H.; Smith, P. L.; Sturgis, S.; Yiv, S. H.; Owen, A. B.; Pharm. Res. 1996, 13, 210.
- 24. Constantinides, P. P.; Pharm. Res. 1995, 12, 1561.
- 25. Dalmora, M. E.; Dalmora, S. L.; Oliveira, A. G.; Int. J. Pharm. 2001, 222, 45.
- 26. Dalmora, M. E.; Oliveira, A. G.; Int. J. Pharm. 1999, 184, 157.
- 27. Friberg, S. E.; Bothorel, P.; Microemulsions: Structure and dinamics, CRC: Boca Raton, 1988.
- 28. Ho, H.; Hsiao, C. C.; Sheu, M. T.; J. Pharm. Sci. 1996, 85, 138.
- 29. Lawrence, M. J.; Eur. J. Drug Metab. Pharmacokinet. 1994, 3, 257.
- 30. Hoar, T. P.; Schulman, J. H.; Nature 1943, 152, 102.
- 31. Schulman, J. H.; Stoeckenius, W.; Prince, L. M.; J. Phys. Chem 1959, 63, 1677.
- 32. Actis, G. C.; Vopes, R.; Rizzetto, M.; Eur. J. Gastroenterol. Hepatol. 1999, 8, 905.
- 33. Shah, D. O., ed.; Micelles, microemulsions, and monolayers: Science and technology; Marcel Dekker: New York, 1998.
- 34. Berthod, A.; J. Chim. Phys 1983, 80, 407.
- 35. Garti, N., ed.; Thermal behavior of dispersed systems; Marcel Dekker: New York, 2001.
- 36. Thevenin, M. A.; Grossiord, M. C.; Poelman, M. C.; Int. J. Pharm. 1996, 137, 177.
- 37. Khan, A.; Curr. Opin. Colloid Interface Sci. 1996, 5, 614.
- 38. Rosoff, M. Em Pharmaceutical dosage forms: Disperse systems; Lieberman, H. A.; Martin, M. R.; Banker, G. S., eds.; Marcel Dekker: New York, 1998, vol. 3.
- 39. Hellweg, T.; Curr. Opin. Colloid Interface Sci. 2002, 7, 50;
- Hiragi, Y.; Sano, Y.; Matsumoto, T.; J. Synchroton Radiat. 2003, 1, 193;
- Kang, S. Y.; Seong, B. S.; Jung, H. T.; Biomacromolecules 2003, 4, 360;
- Steyler, D. C.; Robinson, B. H.; Eastoe, J.; MacDonald, I. Em Micelles, microemulsions, and monolayers: Science and technology; Shah, D. O., ed.; Marcel Dekker: New York, 1998.
- 40. Binks, B. P.; Cho, W-G.; Fletcher, P. D. I.; Tetsev, D. N.; Langmuir 2000, 16, 1025.
- 41. Jacovwics, J.; Berthiaume, M. D.; Cosmetics & Toiletries 1993, 108, 65.
- 42. Constantinides, P. P.; Lancaster, C. M.; Marcello, J.; Chiossome, D. C.; Orner, D.; Hidalgo, I.; Smith, P. L.; Sarkahian, A. B.; Yiv, S. H.; Owen, A. J.; J. Controlled Release 1995, 34, 109.
- 43. Swenson, E. C.; Curatolo, W. J.; Adv. Drug Delivery Rev. 1992, 8, 39.
- 44. Maranhão, R. C.; Garicochea, B.; Silva, E. L.; Llacer-Dorlhiac, P.; Cadena, S. M. S.; Coelho, I. J. C.; Meneghetti, J. C.; Pileggi, F. J. C.; Chamone, D.; Cancer Res. 1994, 54, 4660.
- 45. Maranhão, R. C.; Graziani, S. R.; Brandizzi, L. L.; Coelho, I. J.; Oliveira, M. A.; Hegg, R.; Pinotti, J. A.; Pilleggi, F.; Meneguetti, C. A.; Can. J. Infect. Dis. 1995, 6, 227.
- 46. Maranhão, R. C.; Hungria, V. T. M.; Chamone, D. A. F.; Chianote, C. S.; Int. J. Hematol. 1996, 64, 127.
- 47. Maranhão, R. C.; Roland, Y.; Ramiures, J.; Gonçalves, R. P.; Mesquita, C. H.; Pileggi, F.; Lipids 1997, 32, 627.
- 48. Owens, D. M.; Baillie, G.; Halbert, W. G.; Int. J. Pharm. 2001, 228, 109.
- 49. Correa, M. A.; Tese de Doutorado, Universidade de São Paulo, Brasil, 1997.
- 50. Dalmora, M. E.; Dissertação de Mestrado, Universidade Federal de Santa Maria, Brasil, 1997.
- 51. Takeo, M.; Disperse systems, Willey-VCH: New York, 1999.
- 52. Gasco, M. R.; Pattarino, F.; Lattanzi, F.; Int. J. Pharm. 1990, 62, 119.
- 53. Paul, B. K.; Moulik, S. P.; J. Dispersion Sci. Technol. 1997, 18, 301.
- 54. Sarciaux, J. M.; Acar, L.; Sado, P. A.; Int. J. Pharm. 1995, 120, 127.
- 55. Osborne, D. W.; Ward, A. J. I.; O' Neill, K. J.; Drug Dev. Ind. Pharm. 1988, 14, 1202.
- 56. Linn, E. E.; Pohland, R. C.; Byrd, T. K.; Drug Dev. Ind. Pharm. 1990, 16, 899.
- 57. Mitra, S.; Gaur, U.; Ghosh, P. C.; Maitra, N. A.; J. Controlled Release 2001, 74, 317.
- 58. http://home.eznet.net/~webtent/drugmonitor.html, acessada em Novembro 2003.
Datas de Publicação
-
Publicação nesta coleção
09 Mar 2004 -
Data do Fascículo
Fev 2004
Histórico
-
Recebido
25 Set 2002 -
Aceito
27 Jun 2003