Resumo
Though sharing only a short part on the natural products timeline, the studies on marine products has already handed in four new drugs to the clinical arsenal and brought up a long and promising list of unique molecules to pre-clinical and clinical trials. Thus, as the available analytical resources improve and the interest of large pharmaceutical companies arises, medical use of marine products has definitely become a reality.
marine biotechnology; natural products; marine drugs
marine biotechnology; natural products; marine drugs
ARTIGO
Organismos marinhos como fonte de novos fármacos: histórico & perspectivas
Marine organisms as a source of new pharmaceuticals: history and perspectives
Letícia Veras Costa-Lotufo* * e-mail: lvcosta@secrel.com.br , I; Diego Veras WilkeI; Paula Christine JimenezI; Rosângela de A. EpifanioII
ILaboratório de Oncologia Experimental e Instituto de Ciências do Mar, Universidade Federal do Ceará, Rua Cel. Nunes de Melo 1127, 60430-270 Fortaleza - CE, Brasil
IIInstituto de Química, Universidade Federal Fluminense, Campus do Valonguinho, 24020-150 Niterói - RJ, Brasil
ABSTRACT
Though sharing only a short part on the natural products timeline, the studies on marine products has already handed in four new drugs to the clinical arsenal and brought up a long and promising list of unique molecules to pre-clinical and clinical trials. Thus, as the available analytical resources improve and the interest of large pharmaceutical companies arises, medical use of marine products has definitely become a reality.
Keywords: marine biotechnology; natural products; marine drugs.
INTRODUÇÃO
Os produtos naturais têm sido a maior fonte de inspiração para diversas áreas da química e da ciência de um modo geral. Usando, copiando ou modificando as moléculas sintetizadas pelos seres vivos, o homem tem obtido inovações para o seu benefício em diversas áreas e, entre elas, a produção de fármacos. Do analgésico morfina, ao antibiótico penicilina, passando pelo anticâncer paclitaxel (Taxol®), estima-se que 80% dos fármacos em uso são produtos naturais ou foram inspirados pela natureza.
Existem várias revisões recentes que podem ser consultadas para uma visão da contribuição dos produtos naturais na indústria farmacêutica.1-4 Neste contexto, os organismos terrestres (principalmente micro-organismos e plantas) são os responsáveis por quase a totalidade dessas substâncias, enquanto que os organismos marinhos, apesar de promissores, passaram a ser investigados de maneira sistemática apenas recentemente. Nesta breve revisão comentaremos alguns aspectos históricos dos estudos químicos e farmacológicos realizados com organismos marinhos e suas implicações científicas, como a descoberta de novos produtos naturais com ação em alvos moleculares peculiares, além da utilização desse conhecimento pela indústria farmacêutica no processo de pesquisa e desenvolvimento (P&D) de fármacos.
HISTÓRICO
Mares e oceanos ocupam mais de 2/3 da superfície da Terra e abrigam quase todos os grupos de organismos vivos, incluindo representantes de 34 dentre os 36 filos descritos. Sendo assim, os ecossistemas marinhos podem ser considerados como detentores da maior biodiversidade filética, com potencial biotecnológico associado praticamente ilimitado.5-8
Até os anos 50, este ecossistema escapou do interesse dos cientistas de produtos naturais, principalmente devido ao difícil acesso às suas profundidades. Com o avanço das técnicas e o advento dos equipamentos seguros de mergulho, na década de 70, algas e invertebrados marinhos puderam dar início às suas histórias nas bancadas dos laboratórios de química e farmacologia.7
O pesquisador Werner Bergmann da Universidade de Yale (E.U.A.) foi um dos pioneiros, na década de 50, no estudo dos produtos naturais marinhos. Um dos exemplos históricos no desenvolvimento de fármacos marinhos teve origem no seu trabalho de isolamento dos nucleosídeos espongouridina (1) e espongotimidina (2) da esponja Tethya crypta (Cryptotethya crypta).9 Análogos sintéticos, como a adenina-arabinosídeo (ara-A) (3) e a citosina-arabinosídeo (ara-C) (4), discutidos a seguir, são até hoje utilizados na terapêutica.10,11

No entanto, apenas após a descoberta de grandes quantidades de prostaglandinas (5ab) no octocoral Plexaura homomalla, em 1978, por Alfred J. Weinheimer e Robert L. Spraggins da Universidade de Oklahoma (E.U.A.), foi que o interesse e o investimento de indústrias farmacêuticas em laboratórios de pesquisa de produtos naturais na busca de novos fármacos de origem marinha foram despertados.12

O estudo de produtos marinhos, pelos 20 anos que se seguiram, respondeu com uma excitante explosão de moléculas peculiares, trazendo estruturas complexas até então desconhecidas, além de potentes atividades biológicas e interações com alvos celulares inusitados.10,13
Nos 10 anos seguintes veio a calmaria, resultante da falta de interesse da indústria farmacêutica, que, à época, preferiu apostar na síntese e na química combinatória para a descoberta de novos fármacos.14 Nesta altura, nenhum fármaco oriundo do mar tinha alcançado as prateleiras das farmácias. Hoje, e desde o início do século corrente, vem-se testemunhando o que pode configurar o renascimento do campo dos produtos naturais marinhos.15-23
Os avanços nas técnicas de isolamento e caracterização química, os progressos e a sofisticação dos bioensaios, que se mostram cada vez mais específicos e acurados, receberam, ainda, o reforço de mais um importante grupo de profícuos produtores de moléculas interessantes: os micro-organismos marinhos.6,24-26 Quase a metade (45%) dos produtos naturais de micro-organismos marinhos foi isolada após o ano 2000, demonstrando o interesse recente nesta área.22 Os micro-organismos marinhos tiveram implicações revolucionárias nos estudos com produtos naturais, não apenas pela diversidade química mas, principalmente, pela perspectiva de sustentabilidade, associada à possibilidade de fermentação para produção de material suficiente para as etapas pré-clínicas e clínicas do desenvolvimento de fármacos.27
A Figura 1 resume a evolução no isolamento de produtos naturais marinhos ao longo dos últimos cinquenta anos. Apesar das cerca de 18.500 substâncias isoladas entre 1965 e 2006, calcula-se que menos de 3% do total estimado de organismos marinhos já tenha sido estudado.23
Além de suas estruturas peculiares, produtos naturais marinhos possuem uma extraordinária diversidade de alvos moleculares com seletividade marcante, o que aumenta sobremaneira o potencial farmacológico e terapêutico dessas moléculas (ver exemplo das conotoxinas, discutido a seguir).10,28
Considerando apenas os protótipos em fase de testes clínicos, podemos reconhecer alguns alvos terapêuticos: canais iônicos, enzimas, microtúbulos, DNA, lisossomos, calmodulina, proteossomos, além de indução de estresse oxidativo e modulação do sistema imunológico.10,28 A grande maioria dos alvos identificados apresentam-se como relevantes no tratamento do câncer e é exatamente no estudo e na terapêutica dessa doença que podemos visualizar o maior impacto das substâncias de origem marinha.10,29,30
ESTADO DA ARTE: A INDÚSTRIA FARMACÊUTICA E OS FÁRMACOS DE ORIGEM MARINHA COMERCIALIZADOS
Todo o conhecimento acumulado do estudo, principalmente no meio acadêmico, de produtos naturais marinhos ressalta o potencial terapêutico dessas moléculas.
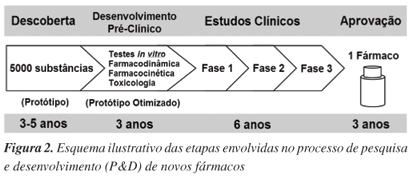
A indústria farmacêutica representa o setor investidor principal das etapas de ensaios pré-clínicos e clínicos no processo de P&D de fármacos, o que, de certa forma, determina quais moléculas têm potencial para atingir o mercado.31,32 Os custos envolvidos nesse processo são extremamente elevados, podendo envolver gastos da ordem de 2 bilhões de reais (900 milhões de dólares), no caso de fármacos anticâncer.33,34 Além disso, o processo é muito demorado, levando cerca de 15 anos da descoberta da molécula à prateleira da farmácia (Figura 2).33,34 Como mencionado anteriormente, a contribuição dos produtos naturais nesse processo é marcante, visto que a maioria das novas entidades químicas introduzidas no mercado para tratamento de doenças tem, em última instância, origem natural.
Se as grandes indústrias farmacêuticas haviam perdido o interesse pelos produtos naturais marinhos, elas estão começando a recuperá-lo, ainda que com alguma cautela, na forma de colaborações com pequenas companhias ou laboratórios institucionais. Uma análise do pipeline das grandes indústrias farmacêuticas mostra que a maioria delas tem investido no desenvolvimento de fármacos baseados em produtos naturais marinhos.35 Vale mencionar que 13 novos fármacos relacionados a produtos naturais foram aprovados entre 2005 e 2007, sendo que cinco representam novas classes de fármacos, incluindo os produtos marinhos trabectedina (Yondelis®) e ziconotídeo (Prialt®).3,4
Nos últimos anos algumas companhias foram fundadas com o objetivo de desenvolver novos fármacos com protótipos de origem marinha. É o caso da PharmaMar, fundada em 1986 e, hoje, uma divisão do grupo Zeltia, que nos últimos 20 anos investiu mais de 1,2 bilhão de reais (420 milhões de euros) na pesquisa em fármacos de origem marinha com potencial para tratamento do câncer. A PharmaMar conta com uma coleção de mais de 65.000 organismos marinhos, de onde já foram isoladas 700 novas entidades químicas e 30 novas famílias de substâncias já foram descritas. Vale ressaltar que atualmente existem 3 substâncias em fase pré-clínica e 5 em fase clínica de testes, sendo que a trabectedina (ET-743, Yondelis®) foi recentemente aprovada pela Comissão Européia para o tratamento de sarcomas de tecidos moles em pacientes refratários a antraciclinas e ifosfamida.36
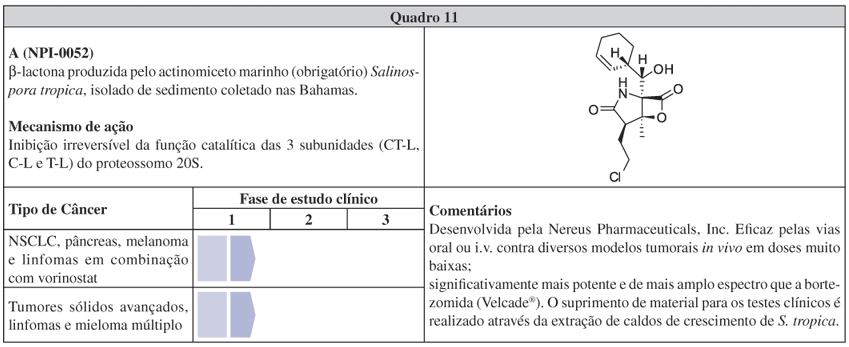
Outra companhia fundada sob a inovadora prerrogativa de prospecção de fármacos a partir de micro-organismos marinhos é a Nereus Pharmaceuticals.37 Fundada em 1998, já possui 2 protótipos em fase 1 de testes clínicos para o tratamento de câncer, incluindo a salinosporamida A (ver Quadro 11), que carrega o trunfo de ter sido a substância a percorrer mais rapidamente a fase de testes pré-clínicos: desde a sua descoberta até o início dos testes clínicos contam-se apenas 3 anos.38,39
Atualmente já existem 4 fármacos relacionados a substâncias de origem marinha em uso clínico - como anticâncer [ara-C (Citarabina®) e trabectedina (Yondelis®)], antiviral [ara-A (Vidarabina®)] e analgésico neuropático [ziconotídeo (Prialt®)].
Ara-C (Citarabina®) e Ara-A(Vidarabina®)
Como mencionado anteriormente, os nucleosídeos isolados por Bergmann e Feeney9 inspiraram a síntese de moléculas como a citosina arabinosídeo [4, ara-C (Citarabina®)] e a adenina arabinosídeo [3, ara-A (Vidarabina®)], fármacos com ações anticâncer e antiviral, respectivamente.40,41 Ara-C (4) é usada principalmente na terapia da leucemia aguda não linfoblástica, enquanto que ara-A (3) é utilizada na forma de colírio para tratamento de infecções oftalmológicas com o vírus Herpes simplex.42 De acordo com Newman e Cragg,10 o grande impacto da descoberta dessas moléculas foi demonstrar, pela primeira vez, a ocorrência de nucleosídeos naturais bioativos contendo açúcares diferentes da ribose e da deoxirribose. Pode-se dizer ainda que essa constatação deu origem a toda uma geração de nucleosídeos incomuns e não naturais com potencial terapêutico, incluindo o fármaco anti-HIV, azidotimidina ou AZT.43
Vale ressaltar que é exatamente nessa característica que reside o mecanismo de ação dessas substâncias que "enganam" a maquinaria de divisão celular e, uma vez incorporadas ao DNA durante a replicação, levam à inibição do processo de síntese e consequente bloqueio da proliferação.44
Trabectedina (Ecteinascidina-743, ET-743, Yondelis®)

Trabectedina (6, ET-743, Yondelis®) é um alcalóide tetra-hidroquinolínico isolado da ascídia caribenha Ecteinascidia turbinata e cujo mecanismo de alquilação de DNA difere da grande maioria dos outros agentes alquilantes. Esta molécula liga-se à guanina em sequências específicas de bases nas fendas menores da dupla hélice, causando uma dobra nas fitas de DNA.45,46 Este dobramento tem diversas consequências celulares que são refletidas na desorganização do citoesqueleto, bloqueio da divisão celular e interferência no reconhecimento e ligação normal de fatores de transcrição ou proteínas ligantes ao DNA, causando ativação ou inibição de determinados genes.47,48 A inibição da transcrição do gene MDR1 é um dos principais e mais relevantes efeitos desta substância, sendo este gene responsável pela produção de glicoproteína-P, uma proteína que realiza o bombeamento de diversos quimioterápicos para fora das células e pode estar superexpressa em células tumorais. A glicoproteína-P confere às células tumorais um perfil de resistência a múltiplas drogas (MDR), o que pode vir a ser prevenido pelo tratamento com trabectedina.49,50
A via de reparo TC-NER (reparo por excisão de nucleotídeos acoplados à transcrição, Transcription Coupled Nucleotide Excision Repair) também parece exercer um papel essencial na atividade da trabectedina.47 Esta via está envolvida no reparo de dano ao DNA induzido por raios UV, determinados carcinógenos e alguns quimioterápicos, como os agentes alquilantes e cisplatina. As células deficientes nesta via de reparo aparecem resistentes ao efeito deste tratamento, o que sugere que esta seja uma via chave na morte celular induzida por trabectedina. Foi demonstrado que as proteínas produzidas pelas células TC-NER proficientes induzem quebras em DNA de fita simples, enquanto que as proteínas mutadas são menos tóxicas às células.51-54
Os primeiros ensaios clínicos com trabectedina foram complicados devido à severa toxicidade e algumas mortes. Entre as principais reações adversas, podem ser citadas neutropenia, transaminite e, mais gravemente, rabdomiólise. Consequentemente, vários estudos foram realizados para o entendimento das reações relacionadas à toxicidade e para o ajuste de doses, o que atrasou a sua progressão normal pelos ensaios clínicos. Entretanto, as modificações adotadas melhoraram substancialmente o perfil toxicológico do fármaco, com a grande vantagem de induzir apenas uma mielotoxicidade residual nas doses terapêuticas.55
Trabectedina foi o segundo protótipo desenvolvido pela PharmaMar e o primeiro fármaco a ser aprovado para uso clínico no tratamento de sarcoma de tecidos moles, tendo recebido autorização da agência EMEA (Agência Européia de Medicamentos, European Medicine Agency) para a comercialização pela União Européia em setembro de 2007.56 Atualmente, segue cumprindo outros protocolos de ensaios clínicos e encontra-se em fase 3 para tratamento de câncer de ovário e em fase 2 para câncer de próstata e mama, osteossarcoma e tumores sólidos pediátricos.57
A Ecteinascidia turbinata foi cultivada com sucesso, mas, para os testes clínicos, o detentor do prêmio Nobel de Química de 1990, E. J. Corey, desenvolveu, em colaboração com outros químicos, um método eficiente de síntese que, patenteado por Harvard, foi licenciado à PharmaMar. Sua produção é hoje realizada em 4 etapas a partir da cianosafracina B, proveniente da cultura da bactéria Pseudomonas fluorescens.58 O sucesso da aquacultura e do processo de síntese do ET-743, sem dúvida, foi um dos fatores que agilizaram a realização dos testes clínicos e sua consequente aprovação como fármaco.
Prialt® (ziconotídeo, ω -conotoxina MVIIA)

O ziconotídeo (7) ou ω-conotoxina MVIIA (Prialt®) é um peptídeo analgésico de 25 aminoácidos isolado do molusco Conus magnus na década de 80.59,60 Como outros peptídeos, o ziconotídeo é sintetizado a partir de aminoácidos naturais.
Utilizadas para paralisar suas presas, as toxinas de Conus têm sido estudadas para diversas aplicações na medicina e são um exemplo da diversidade de alvos encontrada para os produtos de origem marinha e de sua seletividade. Estima-se que cada espécie contenha até 200 diferentes peptídeos biologicamente ativos que se ligam em canais iônicos dependentes de voltagem ou ativados por ligantes externos.61-65 Como exemplos, podemos citar as α-conotoxinas que inibem competitivamente receptores nicotínicos com uma seletividade impressionante entre os seus diversos subtipos, enquanto que as ψ-conotoxinas atuam como inibidores não-competitivos nesse mesmo alvo.66 As σ-conotoxinas antagonizam receptores de serotonina 5-HT3, enquanto as conopressinas são agonistas de receptores de vasopressina.67,68 Com relação aos canais iônicos dependentes de voltagem, as ω-conotoxinas bloqueiam seletivamente diferentes subtipos de canais de cálcio, as κ-conotoxinas bloqueiam canais de potássio, enquanto que as µ-conotoxinas bloqueiam canais de sódio.28,63,65,69
Com sua seletividade com relação ao alvo molecular, o ziconotídeo bloqueia, em concentrações picomolares, o canal de cálcio do tipo N das fibras nociceptivas tipo A-δ e tipo C nas lâminas I e II da raiz dorsal do cordão nervoso de mamíferos, apresentando analgesia em vários modelos de nocicepção.70,71
O extraordinário potencial antinociceptivo das ω-contoxinas (o ziconotídeo é mil vezes mais potente do que a morfina) emerge de duas propriedades: a particular sensibilidade das fibras tipo C a essas substâncias, como mencionado, e a ausência de tolerância ao tratamento com esses peptídeos. Em contraste com os opioides, que são agonistas de receptores acoplados à proteína G e que levam à diminuição no número de receptores após tratamento continuado e à consequente dessensibilização, o tratamento com ziconotídeo não induz tolerância após uso crônico.63,69,72
O primeiro teste clínico com ziconotídeo (Prialt®) em pacientes com dor severa teve início em 1995, sendo que somente em 2004 a agência norte-americana FDA (agência dos E.U.A. para controle e aprovação de medicamentos e alimentos, Food and Drug Administration) aprovou este fármaco de administração intratecal no controle da dor severa, incluindo aqueles pacientes refratários a tratamentos com opioides.72,73 Esse medicamento é comercializado pela Elan Pharmaceuticals.74
PROTÓTIPOS EM ESTUDOS CLÍNICOS: DESAFIOS, SOLUÇÕES E PERSPECTIVAS
Apesar do reduzido número de fármacos de origem marinha, existe um número considerável de substâncias em fase de testes pré-clínicos e clínicos para tratamento de doenças, o que ressalta o potencial destas moléculas na cura de doenças. Durante o processo de P&D de fármacos, de cada 5000 substâncias que entram em fase de testes pré-clínicos apenas 5 passam aos estudos clínicos e uma só molécula resulta num medicamento.34 Embora o grande gargalo da evolução dos estudos com as substâncias de origem marinha tenha sido o suprimento de quantidades adequadas para os testes em humanos, o fator limitante da utilização na terapêutica reside na toxicidade associada ao uso.29,34
De fato, a função ecológica das moléculas farmacologicamente ativas isoladas de fontes marinhas parece ser a de assegurar o sucesso do hospedeiro na competição por espaço ou na defesa contra predadores ou micro-organismos patogênicos, tanto que foram os animais sésseis ou com reduzida capacidade de locomoção, de corpo macio e desprovidos de estruturas físicas de defesa, como esponjas não-calcáreas, ascídias, corais moles, algas e alguns grupos de moluscos sem concha, que cederam grande parte das moléculas que estão, atualmente, em estudos pré-clínicos ou clínicos.15,75 A bioatividade das moléculas de origem marinha, não raro, é extremamente potente, o que reforça a hipótese da sua função protetora, já que elas devem superar a incrível capacidade diluente da água do mar para alcançar o seu alvo e surtir efeito.30 Entretanto, no processo de transformação das "armas químicas" dos organismos marinhos em fármacos para uso humano, essa particularidade repercute na alta toxicidade destas moléculas ou em intoleráveis efeitos colaterais. O baixo rendimento do material, a frequente complexidade estrutural dos produtos naturais e sua consequente dificuldade de síntese somam-se ao rol dos motes que abalizam o desenvolvimento de fármacos de origem marinha.
A história das substâncias que estão ou passaram pela fase de testes clínicos, de certa forma, ilustra os diferentes aspectos relacionados ao desenvolvimento de fármacos a partir de fontes marinhas. A geração de moléculas análogas ou congêneres para avaliar as relações entre estrutura e atividade e as modificações estruturais para otimizar os parâmetros farmacocinéticos e farmacodinâmicos são recursos geralmente disponíveis para a superação da questão da toxicidade.30
Quando se trata de amostrar material para meros estudos químicos ou de screening farmacológico, a convenção rege que a coleção entre 500 e 1000 g (considerando o peso úmido) de cada organismo é uma quantidade bastante razoável e suficiente para cumprir todas as etapas da prospecção sem causar impacto ecológico. Entretanto, quando se chega à molécula bioativa isolada e se vislumbra avançar nos estudos pré-clínicos e, principalmente, nas etapas clínicas, deve-se considerar alternativa outra que a coleta do animal selvagem.15,34
A questão do suprimento de material assombrou o desenvolvimento da área de produtos naturais marinhos desde os seus dias mais remotos. A discussão acerca deste entrave era mais frequente em revisões sobre produtos marinhos publicadas até por volta da virada do século corrente que, à altura, era bastante pertinente.5,15,76-78 O que se experimenta nos dias de hoje já são fases de superação deste aspecto, seja pelo avanço de métodos de síntese e aquacultura ou pelos avanços da biologia molecular, que viabilizou a utilização de bactérias geneticamente modificadas na produção de substâncias de estrutura mais complexa.7,35
A cultura de células dissociadas dos organismos hospedeiros parecia uma solução óbvia para a produção das moléculas bioativas, já que esta técnica é empregada com maestria na produção em larga escala de diversas biomoléculas, como anticorpos e hormônios, usando reatores biológicos. Experimentos com esponjas marinhas chegaram a ser conduzidos durante mais de uma década e atraíram o interesse de grupos de ponta da área de produtos naturais marinhos.79 Entretanto, após muito investimento dispensado para a implementação das linhagens imortais e de alguns casos bem sucedidos nas tentativas de aumentar a concentração intracelular natural da molécula de interesse,80-82 não foi possível expandir este modelo para culturas de maior porte.
Enquanto isso, os argumentos já pouco favoráveis à cultura de células de esponjas para a obtenção de moléculas bioativas foram reforçados pelas crescentes evidências de que muitos dos metabólitos isolados dos invertebrados marinhos seriam, na verdade, produzidos por micro-organismos associados. Sem dúvida, a simbiose ou o comensalismo que plantas e animais marinhos mantêm com vários micro-organismos é uma relação ecológica bem conhecida e amplamente documentada. Os micro-organismos marinhos desenvolveram um aparato metabólico bastante peculiar que é, de certo modo, muito mais promíscuo que o de seres multicelulares. Essa fenomenal capacidade adaptativa não só garantiu a sua sobrevivência em ambientes às vezes extremos, mas também proporcionou um recurso para a produção de metabólitos nunca antes observados para organismos terrestres.83,84 Isso explica muita coisa. Ora, assim como já foram isoladas substâncias quimicamente semelhantes a partir de organismos filogeneticamente distantes, outras originadas por vias biossintéticas completamente distintas achavam-se presentes numa mesma espécie.85
O potencial dos micro-organismos marinhos na busca de novos fármacos é também fundamentado na importância histórica dos micro-organismos terrestres como fonte de fármacos. A rigor, algum esforço já foi empreendido neste sentido e inúmeros metabólitos secundários interessantes já foram isolados a partir desses grupos, mas presume-se que ainda haja um desmesurado número de substâncias por descobrir.86 E, embora sejam necessárias técnicas diferenciadas, muitos dos princípios que embasam a fermentação industrial podem ser aplicados para a obtenção dos metabólitos secundários em larga escala, o que, em tese, resolveria a questão do suprimento de material.87,88
No entanto, para os protótipos ainda em fase de ensaios clínicos ou fármacos já aprovados, a síntese química ainda é o recurso de escolha da indústria farmacêutica para obter a quantidade de material necessária para atender à demanda do mercado. Além do mais, a síntese garante a independência da indústria frente à vulnerabilidade dos recursos biológicos. Entretanto, a complexidade estrutural da maioria dos produtos naturais marinhos acaba por dificultar a elaboração de um processo comercialmente viável.7,34,79 A grande saída para as moléculas de arquitetura mais complexa é investir na elucidação do mecanismo de ação, determinar o grupo farmacofórico e tentar a síntese de moléculas mais simples, de bioatividade comparável e, quem sabe até, com menor toxicidade. Esse processo é chamado "simplificação estrutural" ou síntese "orientada pela função".89,90 Aliás, com os avanços nas técnicas de síntese e modelagem molecular, a tendência é que a estrutura original dos produtos naturais passe a servir principalmente como a base para o desenvolvimento de fármacos ao invés de serem eles próprios o princípio terapêutico.3,39,91,92
A produção de análogos a partir de estruturas líder como a didemnina B, dolastatinas, halichondrina B e briostatina são exemplos de protótipos que sofreram modificações para facilitar ou melhorar o rendimento da sua síntese ou reduzir a toxicidade sem experimentar diminuição da sua potência. De qualquer sorte, algumas moléculas de ocorrência natural que estão percorrendo os ensaios clínicos foram bem sucedidas nas tentativas de síntese total ou parcial, entre elas a trabectedina, ω-conotoxina, aplidina, discodermolido, kahalalido F e espisulosina.
A didemnina B (Quadro 1), um depsipeptídeo cíclico isolado da ascídia caribenha Trididemnun solidum,93 é considerada efetivamente a primeira substância natural marinha a entrar para testes clínicos, nesse caso, como um agente anticâncer. Nos estudos in vitro e in vivo, esta substância demonstrou uma potente atividade antiviral, imunossupressora e anticâncer, surpreendendo com a CI50 de 1 nM, em média, e com uma notável atividade antitumoral contra leucemias e melanomas.15,93 O mecanismo de ação da didemnina B é complexo e ainda está por ser completamente esclarecido. Independentemente, os resultados animadores dos estudos com linhagens de células tumorais e camundongos com tumores transplantados foram razão suficiente para iniciar os ensaios clínicos, em 1986.94,95
Apesar dos inúmeros e variados esquemas de tratamento propostos e dos diversos tumores experimentados, a didemnina B mostrou ser por demais tóxica, também desapontando no quesito eficácia, para prosseguir com os ensaios clínicos, que foram interrompidos na fase 2 em meados da década de 90. Alguns autores argumentam que a logística envolvida no ajuste de doses e nos esquemas de administração utilizados não teria sido otimizada para favorecer a atividade antitumoral e adequava-se melhor aos protocolos de estudo de agentes imunomoduladores.95 Seja como for, a didemnina B foi retirada dos testes clínicos deixando uma preciosa contribuição para a formação de químicos e farmacologistas básicos e clínicos da área de produtos naturais marinhos.
Todavia, este ainda não é o final da história. A aplidina (Quadro 1), isolada da ascídia mediterrânea Aplidium albicans, é um depsipeptídeo cíclico similar à didemnina B e também sabida por de-hidrodidemnina B.96 A aplidina demonstrou ser a mais ativa num estudo relacionando estrutura e atividade antiviral e citotóxica de uma série de 42 didemninas análogas naturais ou sintéticas, entre as quais a didemnina B.97 Em modelos in vivo, a aplidina conseguiu reduzir drasticamente o volume de diversos tumores, inclusive daqueles resistentes à didemnina B, enquanto demonstrava uma significativa melhoria da toxicidade associada ao uso.98 Seu mecanismo de ação é bastante peculiar e envolve várias vias que levam à apoptose, mas ainda não está completamente esclarecido. Sabe-se que seus efeitos implicam principalmente no bloqueio da divisão celular (devido à inibição da síntese de DNA e proteínas) e no enfraquecimento da angiogênese tumoral (pela diminuição da secreção de VEGF e da expressão do receptor VEGF-r1 e à inibição da enzima ornitina-descarboxilase).99-101
A aplidina foi licenciada para a PharmaMar sob o registro de Aplidin® (plitidepsina) e ingressou nos testes clínicos em 1999, vindo a se beneficiar diretamente das informações geradas nos estudos com didemnina B. Interessantemente, a simples oxidação do radical 2-hidroxipropanoil a 2-oxopropanoil transforma substancialmente o seu perfil toxicológico.10,30 De modo geral, o tratamento com aplidina tem sido bem tolerado e a toxicidade neuromuscular, seu principal fator limitante, pode ser contornada com a coadministração de L-carnitina.102 Vale ressaltar que reações tipicamente associadas à quimioterapia do câncer, como mucosite, alopecia e mielossupressão, são incomuns neste tratamento, que se encontra em fase 2 para uma variedade de tumores sólidos e hematológicos como terapia única e em fase 1 para terapia combinada com outros quimioterápicos e para o tratamento de leucemias pediátricas agudas .57,103
Para os diversos kahalalidos (A-Q), outra série de depsipeptídeos, já foram registradas suas atividades anticâncer, antimalarial, antipsoríase, tuberculostática, antifúngica e antiviral, mas foi apenas o kahalalido F (KF, Quadro 2) que apresentou significativa atividade contra linhagens de células tumorais humanas, inclusive pulmão e cólon, e contra alguns micro-organismos comumente oportunistas de pacientes portadores de HIV.104-106
KF atua na membrana dos lisossomos, alterando sua função basal e induzindo a morte celular preferencialmente por oncose,107-109 um processo mais típico, mas não restrito, a células do tecido ósseo, que geralmente é resultante de uma isquemia celular devido à falha no bombeamento iônico que pode ser causada por agentes tóxicos que bloqueiam a geração de ATP ou aumentam a permeabilidade da membrana plasmática, e que evolui, finalmente, para um processo de necrose.110 A atividade citotóxica de KF independe de p53 ou da expressão do perfil MDR e está correlacionada à expressão basal de Erb-B3.109
Durante a fase 1 dos ensaios clínicos, KF foi avaliado em câncer de próstata andrógeno-refratário e apresentou-se bastante tolerável e sem efeitos adversos sérios. Atualmente, o suprimento de kahalalido F é viabilizado através de síntese e esta molécula encontra-se na fase 2 de testes clínicos para o tratamento de carcinoma hepatocelular, NSCLC (câncer de não pequenas células de pulmão, Non-Small Cell Lung Cancer) e melanoma maligno avançado, além de estar sendo avaliado para o tratamento de psoríase severa.36
Outra classe de peptídeos importantes são as dolastatinas naturais e seus derivados (Quadro 3), peptídeos lineares encontrados em pequenas quantidades no molusco Dolabella auricularia e que agem em concentrações picomolares em diversos tipos de células tumorais.111,112 As dolastatinas ligam-se à tubulina no sítio dos alcalóides da Vinca, mas em um domínio diferente, o que gerou o entusiasmo para início dos estudos clínicos.113,114 A síntese da dolastatina 10, a mais potente da série, possibilitou o ingresso desta molécula nos estudos clínicos de fases 1 e 2 pelo NCI (National Cancer Institute - Instituto Nacional do Câncer dos Estados Unidos). Embora a toxicidade tenha sido tolerada nas doses administradas na maioria dos casos, os estudos clínicos, já em fase 2, cessaram de fato por ausência de resultados significativos.115 A partir de um estudo estrutura-atividade, foram desenvolvidos uma série de análogos com o objetivo de manter ou melhorar a citotoxicidade e reduzir a toxicidade. Atualmente 3 derivados sintéticos das dolastatinas encontram-se em estudos clínicos contra vários tipos de tumores.
O TZT-1027, também conhecido por soblidotina ou auristatina (Quadro 3), é um análogo sintético da dolastatina 10 licenciado pela Daiichi Pharmaceuticals.30 Após um estudo clínico de fase 1 para determinar a toxicidade em pacientes com tumores sólidos, seguiram-se os estudos de fase 2 para tratamento de NSCLC e sarcoma de tecido mole em estado avançado ou metastático. O TZT-1027 foi considerado seguro e bem tolerado, porém não apresentou atividade antitumoral.116-118
LU-103793 (ou cemadotina) e ILX-651 (ou sintadotina ou tasidotina) são análogos sintéticos da dolastatina 15 desenvolvidos pela BASF Pharma. Nos testes clínicos, a cemadotina (Quadro 3) mostrou-se segura nos estudos de fase 1 e melhorou a qualidade de vida de alguns pacientes com câncer de pulmão em fase 2, porém não demonstrou resultados objetivos contra os tumores.119-127
A sintadotina (Quadro 3) é uma dolastatina de 3ª geração que age estabilizando microtúbulos, porém com um mecanismo singular, distinto das outras dolastatinas ou mesmo dos taxanos. O ILX-651 traz grandes vantagens, como ser ativo por via oral e possuir uma janela terapêutica maior que a da cemadotina. A atividade em modelos xenográficos com tumores resistentes à terapia com taxanos também foi muito apreciada porque, não raro, pacientes com melanoma desenvolvem resistência à quimioterapia.128-130 Em 2000 a ILEX comprou os direitos da BASF Pharma sobre a sintadotina, gerando uma expectativa de aceleração dos estudos clínicos devido à expertise desta indústria no desenvolvimento de quimioterápicos anticâncer. Em fase 1, o ILX-651 demonstrou boas respostas, havendo até cura de um paciente com melanoma metastático e estabilização da doença em outros casos. Estes resultados animaram bastante os pesquisadores, sobretudo porque há poucas opções para o tratamento de melanoma metastático. Além do melanoma, também foram tratados tumores sólidos recorrentes e NSCLC, com bons resultados ainda em fase 1.131 Com relação à toxicidade, a sintadotina foi considerada segura e bem tolerada, sendo relatados apenas efeitos colaterais de menor importância como alopecia, náuseas, vômitos e fadiga.128 Os estudos de fase 2 estão em andamento.130
A síntese estereosseletiva dessas substâncias, assim como de outros peptídeos marinhos, é de fundamental importância uma vez que os mesmos possuem aminoácidos incomuns como, por exemplo, D-valina, Z-de-hidroaminobutírico, N,N-dimetilvalina e (2R,3R,4S)- dolaproína. A síntese de peptídeos, relativamente simples para aqueles contendo aminoácidos comuns, é uma técnica já bem desenvolvida pelos químicos. Cabe ressaltar, entretanto, o trabalho de síntese recente em excelente rendimento e estereosseletividade de aminoácidos incomuns dos peptídeos de dolastatinas, recentemente publicado pelo grupo do Prof. Hélio A. Stefani da Universidade de São Paulo.132-134
Entre os policetídeos, o mais importante pela sua história é a briostatina, uma das duas substâncias em fase de testes clínicos que são obtidas diretamente do organismo no seu ambiente natural (briostatina 1 e o Æ-941). A briostatina 1 (Quadro 4), uma lactona macrocíclica, é considerada a molécula mais promissora da sua família tendo sido isolada do briozoário Bugula neritina por Pettit e colaboradores em 1970.135 Devido à grande complexidade, sua elucidação estrutural só foi alcançada na década seguinte ao seu descobrimento.136 Dentre as diversas atividades estudadas, destacam-se a regulação da apoptose, sinergismo com outros quimioterápicos anticâncer e estimulação do sistema imunológico.137-139 Atualmente, o maior interesse dos estudos com a briostatina 1 é na quimioterapia anticâncer, já se contando mais de 80 estudos clínicos de fase 1 ou 2 realizados ou em andamento.140
Os estudos clínicos com briostatina 1 apresentaram, de forma geral, melhores resultados nas abordagens associadas que nos esquemas de monoterapia.30 Possivelmente, o seu efeito modulador da PKC favorece o sinergismo com outros quimioterápicos e acentua a resposta contra os tumores. Alguns exemplos de esquema de estudos clínicos bem sucedidos com associação da briostatina 1 são com: alcalóides da vinca ou ara-C, demonstrando respostas importantes contra leucemias em estudos de fase 1, 50% de resposta objetiva na associação com fludarabina em leucemia linfocítica crônica e linfoma não-Hodgking e, ainda, alta remissão, embora incompleta, com paclitaxel em pacientes com NSCLC em estudos de fase 2.140,141 A mialgia é o principal efeito colateral relatado, mas, na maioria dos casos, pode ser abordada pelas terapias convencionais.142-144
A coleta de B. neritina do ambiente natural não é e nem deve ser uma opção considerada para a obtenção de briostatina 1. O baixo rendimento do processo de extração não seria compatível com a demanda do mercado e, principalmente, o desastroso impacto ambiental tornaria essa prática insustentável. Só para a realização dos estudos clínicos foram coletadas 13 toneladas de B. neritina - que renderam míseros 18 g da molécula! Neste cenário, o NCI financiou um trabalho pioneiro para viabilizar a produção de briostatina 1 a partir do cultivo artificial de B. neritina.138
Embora o cultivo de B. neritina em tanques tenha sido frustrante, a maricultura no campo, i.e., o cultivo em estruturas montadas dentro mar, foi um sucesso.34,145 De cada uma das 200 estruturas obteve-se um rendimento anual aproximado de 200 mg de briostatina 1, o suficiente para tratamento de 130 pacientes com leucemia por um ano. Não obstante, o custo do equivalente-grama de briostatina 1 foi alto, chegando a bater os 140 mil reais (60 mil dólares), sem contar com os gastos com infra-estrutura de cerca de 35 milhões de reais (15 milhões de dólares), extração, purificação e formulação necessários. Ainda assim, essa é a única forma possível de obtenção dessa substância em quantidade suficiente para suprir a demanda da clínica sem causar impactos ambientais negativos. Por outro lado, uma análise custo/benefício favorece a produção de briostatina 1 por aquacultura levando em consideração seu amplo potencial como adjuvante na terapia do câncer e em enfermidades com comprometimento do sistema imunológico.145
Uma interessante observação ecológica resultou numa formidável descoberta que pode vir a ser uma alternativa mais razoável para a obtenção de briostatina 1 que a aquacultura do briozoário hospedeiro: apenas os exemplares de Bugula neritina coletados em profundidades superiores a 9 m apresentavam quantidades detectáveis das briostatinas 1-3, enquanto que estes sinais não eram detectados nos animais de águas mais rasas. As briostatinas guardam grande semelhança com metabólitos secundários bacterianos sintetizados pela via que envolve a enzima policetídeo sintase tipo 1 (PKS-1) e os estudos apoiados em evidências moleculares revelaram que as briostatinas são produzidas pela proteobactéria simbionte Candidatus Endobugula sertula.146,147 Apesar de esta bactéria ainda não ter se rendido aos meios de cultivo, os estudos com suas PKS aventaram, quiçá, outra opção para a produção de briostatinas e derivados por um processo economicamente viável.
Os esforços de alguns grupos de químicos orgânicos têm se concentrado na simplificação e aumento da eficiência da síntese das briostatinas. Recentemente, pesquisadores da Universidade de Stanford, E.U.A., desenvolveram um método mais econômico, constando de apenas 39 etapas para a síntese da briostatina 16, alternativo às iniciais 70 etapas necessárias.148 Mesmo assim, este ainda não é um processo economicamente viável para a produção em larga escala desta classe de substâncias.149-151 No caso da briostatina, o derivado briolog é um exemplo de simplificação molecular e, consequentemente, de sua síntese, em que a atividade farmacológica é mantida.90,152
Já a aquacultura da esponja Discoderma dissoluta, produtora do discodermolido, outro policetídeo, não foi bem sucedida e os esforços se concentram em aprimorar sua síntese, bem como a de seus análogos.153-158 O discodermolido, um policetídeo isolado em 1990 por Gunasekera e colaboradores,159 foi sintetizado e licenciado pela Novartis Pharmaceuticals e entrou em estudos clínicos de fase 1 como agente anticâncer em 2002.
Utilizando uma bactéria produtora de policetídeos geneticamente modificada e suas policetídeos sintases, Burlingame e colaboradores publicaram, em 2006, a síntese de um intermediário-chave quiral na sua preparação. Este é um dos vários exemplos atuais do uso da genômica em algumas etapas do desenvolvimento de fármacos.160,161
O discodermolido é o agente estabilizante de microtúbulos mais potente já descrito, apresentando seus efeitos via uma distintiva parada do ciclo celular seguida de morte por apoptose. Embora o paclitaxel e o discodermolido compartilhem do mesmo mecanismo de ação, o último é eficiente contra linhagens resistentes ao primeiro, tanto in vitro como in vivo.162-164
O mesilato de eribulina (E-7389, Quadro 6) é um análogo sintético de outro policetídeo natural, a halicondrina B. Produzida pela Eisai Co Ltda, possui estrutura mais simples, porém com a mesma potência do produto natural original.165 A baixa toxicidade é característica marcante nos estudos clínicos e, devido aos resultados positivos das fases 1 e 2 contra diversos tipos de tumores, os estudos de fase 3 para câncer de mama já se iniciam com boas expectativas, sustentadas, inclusive, nas indicações pré-clínicas.166,167 A mielossupressão e neutropenia são os efeitos tóxicos de maior relevância, porém a eribulina induz uma neurotoxicidade menos severa e menos frequente que os demais agentes conhecidos que atuam em microtúbulos.167
A espisulosina (Quadro 7), inicialmente isolada do molusco Mactromeris (= Spisula) polynyma em 1999, é hoje produzida através de síntese e encontra-se em fase 1 de ensaios clínicos para pacientes com tumores sólidos, sob licença da PharmaMar.168,36 A sua estrutura se parece muito com a dos esfingolipídeos, lipídeos complexos desprovidos de glicerol onde o álcool é a esfingosina. As células tratadas com espisulosina perdem a organização dos microfilamentos de actina e entram em apoptose por vias diferentes daquelas induzidas pelos esfingolipídeos naturais. Com estas semelhanças, acredita-se que a espisulosina tenha ação em receptores de esfingolipídeos que, por sua vez, modulam a atividade de proteínas RhoA.30,169,170
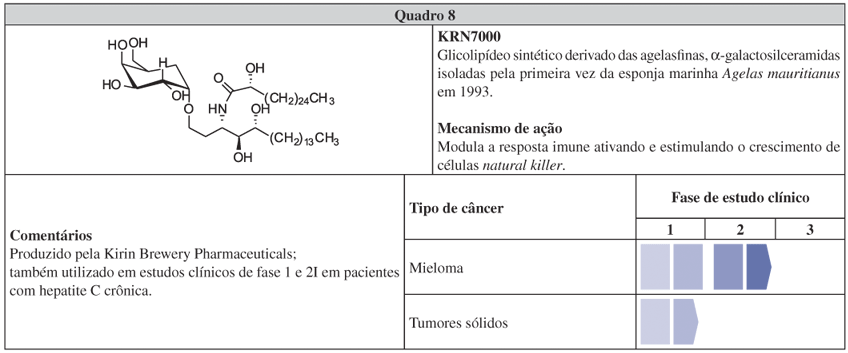
O KRN7000 (Quadro 8) é um análogo das agelasfinas, glicolipídeos isolados da esponja Agelas mauritanus, sob os esforços da Kirin Brewery Pharmaceuticals em 1993.171 Assim como a eribulina, apresenta baixa toxicidade nos estudos clínicos, embora seu mecanismo de ação seja diferente. Essa substância possui uma nova α-galactosilceramida ligada a uma cadeia de ácido graxo longa (C26) e uma esfingosina (C18). Em estágio pré-clínico, foi eficiente em diferentes modelos de metástase172,173 e, nos estudos clínicos de fase 1, não foram observados efeitos colaterais limitantes. Interessantemente, apesar de apenas um pequeno grupo de pacientes ter respondido ao tratamento, esse grupo possuía grande número de células assassinas naturais (denominadas natural killers). Por conta dessa observação, o planejamento dos estudos seguintes considerará uma pré-seleção de pacientes com alta contagem de assassinas naturais.174 A potencial atividade imunoestimulante das agelasfinas já havia sido descrita e, na verdade, o direcionamento dos estudos clínicos de fase 1 desenvolvidos na Europa e Ásia com KRN7000 foi para a imunoterapia do câncer.171,175
Zalypsis® é uma nova entidade química, completamente sintética, mas baseada em produtos naturais marinhos (Quadro 9). Na etapa pré-clínica, Zalypsis® demonstrou uma potente atividade citotóxica in vitro, tendo sido testado contra uma variedade de linhagens celulares humanas derivadas de tumores sólidos ou hematológicos e apresentando CI50 na casa de pico ou nanomolar.176-179 Mais além, Zalypsis® foi igualmente potente contra as linhagens MM1R e RPMI-LR5, selecionadas pela sua resistência aos tratamentos convencionais para mielomas.180 Para os ensaios in vivo, foram utilizados modelos murinos xenográficos de tumores humanos de mama, próstata, estômago, rins e bexiga, apresentando resultados positivos tanto para a atividade antitumoral como na avaliação do perfil toxicológico. A citotoxicidade desta substância ainda não está perfeitamente esclarecida, principalmente porque consiste de vários estágios que envolvem o bloqueio do ciclo celular, induzindo um acúmulo de células nas fases G1/G0, seguido de apoptose. Interessantemente, a citotoxicidade desta substância requer a sua ligação ao DNA, entretanto, esta ligação não ativa o ponto de checagem de "dano em DNA", sugerindo que o seu modo de ação não está ligado a danos à dupla-fita.36,176,180,181
Outro medicamento em potencial cujo suprimento é garantido pela maricultura é o agente antiangiogênico Neovastat® (Æ-941, Quadro 10), desenvolvido pela companhia canadense Æterna Zentaris. Neovastat® não é uma substância isolada, mas um extrato líquido padronizado da fração < 500 kDa da cartilagem do tubarão Squalus acanthias182,183 e que já completou estudos de fase 3 para o tratamento de carcinoma renal metastático e para carcinoma pulmonar de células não-pequenas em combinação com outros quimioterápicos ou com radioterapia.57 O extrato é produzido exclusivamente a partir de tubarões enquadrados taxonomicamente e mantidos em cativeiro sob pomposas condições de sustentabilidade.182 Todavia, a problemática do suprimento voltou à tona neste caso em que não se reconhece qualquer outra possibilidade de obtenção do material e, considerando o alastramento do uso de Neovastat® com a comercialização, a exploração a longo prazo de espécies de tubarão cujas populações naturais já estão ameaçadas é, do ponto de vista ecológico, no mínimo, desaconselhável.
Entre os protótipos de fármacos obtidos de micro-organimos marinhos, o exemplo da β-lactona salinosporamida A (NPI-0052, Sal A, Quadro 11) destaca, com particular evidência, o predicado que seria a "carta na manga" da prospecção de fármacos no ambiente marinho. O actinomiceto marinho-restrito Salinospora tropica, resgatado de sedimento das Bahamas para produzir em laboratório a inusitada molécula, alocou Sal A no pipeline da Nereus Pharmaceuticals Inc.38 Seu mecanismo de ação consiste numa potente e seletiva inibição da função catalítica dos proteossomos, estruturas celulares responsáveis pela degradação de proteínas.184 Em 2003, os proteossomos foram validados enquanto alvo para a quimioterapia do câncer com a aprovação da bortezomida (Velcade®) para o tratamento de mieloma múltiplo.185,186 Atualmente, Sal A está passando pelo ajuste de dose em alguns ensaios simultâneos de fase 1.57 Apesar de esta molécula ter atraído considerável atenção por parte dos químicos sintéticos por sua densa funcionalidade e baixo peso molecular,187,188 é de extrema relevância ressaltar que o suprimento de Sal A para os testes clínicos é completamente obtido através da extração de caldos de crescimento de uma linhagem de S. tropica. Em outras palavras, é o produto natural em si que está percorrendo os testes clínicos e não o produto sintetizado, apresentando a conjuntura em que a fermentação teve o melhor custo/benefício para a obtenção do princípio ativo. Com os ajustes realizados para superar os efeitos corrosivos da fermentação salina e da instabilidade do anel β-lactona em solução aquosa, juntamente com a otimização das condições de cultivo e extração, é possível produzir até 450 mg de Sal A por cada L de meio de cultura com grau de pureza >98% (o que é adequado para a realização dos testes clínicos e para a produção comercial).38
NPI-2358 (Quadro 12) é uma molécula que também está sendo desenvolvida pela Nereus Pharmaceuticals Inc. Esta dicetopiperazina é um dos cerca de 200 análogos sintéticos obtidos a partir da halimida (NPI-2350), um produto natural isolado de uma espécie de Aspergillus sp. de origem marinha.37,57
NPI-2358 liga-se à β-tubulina solúvel no citosol impedindo a sua polimerização, sem, contudo, afetar a dinâmica dos microtúbulos já formados. Os estudos in vivo determinaram uma notória seletividade de NPI-2358 para células endoteliais imaturas em proliferação, aquelas responsáveis pela neovascularização essencial para o suprimento de sangue dos tumores sólidos, mas salvando, entretanto, as células do endotélio maduro. O bloqueio da polimerização da tubulina é iniciado após poucos minutos de exposição à substância, afetando, portanto, forma, adesão e proliferação das células do endotélio imaturo, seguida de um rápido colapso e oclusão da teia vascular tumoral. Finalmente, pela falta de nutrição, as células tumorais são levadas à morte por necrose.189
CONSIDERAÇÕES FINAIS
As conquistas e o progresso da pesquisa de produtos naturais marinhos já fazem honrado o seu lugar na indústria farmacêutica, e a promessa de fármacos de origem marinha tornou-se, definitivamente, realidade. Existe, claramente, um número considerável de substâncias com potencial farmacêutico, traduzindo-se em novas alternativas terapêuticas, principalmente para o tratamento do câncer. Neste contexto, a diversidade química dos produtos naturais de organismos marinhos associada à pluralidade dos alvos terapêuticos oferece perspectivas inovadoras na abordagem clínica das neoplasias malignas. Muitas das substâncias aqui apresentadas atuam em células resistentes a múltiplas drogas. O antimitótico discodermolido, por exemplo, apesar de atuar na tubulina, tal os alcalóides da vinca e os taxanos, não é substrato da glicoproteína-P. Isso significa mais uma frente no combate ao câncer em pacientes que já não respondem à terapia convencional.
Além disso, o desenvolvimento de muitos dos produtos que estão em fase de estudos clínicos está intimamente atrelado ao surgimento de métodos inovadores, particularmente, para garantir o suprimento dessas substâncias através de aquacultura, síntese parcial ou total, incluindo novos derivados com propriedades farmacológicas aprimoradas. Contudo, essa área ainda está longe de atingir a maturidade, e a busca por novas fontes e estratégias de produção em larga escala continua. Bactérias, fungos e cianobactérias também despontam como novas fontes, desafiam microbiologistas, e vislumbra-se na biologia molecular a possibilidade da utilização da metagenômica para viabilizar o estudo dos seus metabólitos. O uso de técnicas de screening in silico e a criação de bibliotecas direcionadas são estratégias para acelerar o processo de descoberta de novas moléculas utilizando sistemas automatizados (high-troughput screening). A clonagem de genes e expressão em bactérias heterólogas já é uma alternativa viável para a síntese de alguns policetídeos. Por fim, as metodologias de síntese que incorporam reações enzimáticas complexas serão fortes aliados na obtenção de quantidades suficientes das moléculas de origem marinha para os estudos clínicos e eventual comercialização.
Recebido em 15/1/09; aceito em 11/3/09; publicado na web em 2/4/09
- 1. Newman, D. J.; Cragg, G. M.; Snader, K. M.; J. Nat. Prod. 2003, 66, 1022.
- 2. Newman D. J.; Cragg, G. M.; J. Nat. Prod. 2007, 70, 461.
- 3. Harvey, A. L.; Drug Discov. Today 2008, 13, 894.
- 4. Butler, M. S.; Nat. Prod. Rep. 2008, 25, 475.
- 5. Munro, M. H. G.; Blunt, J. W.; Dumdei, E. J.; Hickford, S. J. H.; Lill, R. E.; Li, S.; Battershill, C.N.; Duckworth, A. R.; J. Biotech. 1999, 70, 15.
- 6. Cordell, G. A.; Phytochemistry 2000, 55, 463.
- 7. Fenical, W.; Oceanography 2006, 19, 111.
- 8. Lam, K. S.; Curr. Opin. Microbiol. 2006, 9, 245.
- 9. Bergmann, W.; Feeney, R. J.; J. Org. Chem. 1951, 16, 981.
- 10. Newman, D. J.; Cragg, G. M.; Curr. Drug Targets 2006, 7, 279.
- 11. Pinto, A. C.; Silva, D. H. S.; Bolzani, V. S.; Lopes, N. P.; Epifanio, R. de A.; Quim. Nova 2002, 25, 45.
- 12. Weinheimer, A. J.; Spraggins, R. L.; Tetrahedron Lett. 1969, 59, 5185.
- 13. Faulkner, D. J.; Nat. Prod. Rep. 2000, 17, 1.
- 14. Ganesan, A.; Pure Appl. Chem. 2001, 73, 1033.
- 15. Faulkner, D. J.; Nat. Prod. Rep. 2000, 17, 7.
- 16. Faulkner, D. J.; Nat. Prod. Rep. 2001, 18, 1.
- 17. Faulkner, D. J.; Nat. Prod. Rep. 2002, 19, 1.
- 18. Blunt, J. W.; Copp, B. R.; Munro, M. H.; Northcote, P. T.; Prinsep, M. R.; Nat. Prod. Rep. 2003, 20, 1.
- 19. Blunt, J. W.; Copp, B. R.; Munro, M. H.; Northcote, P. T.; Prinsep, M. R.; Nat. Prod. Rep 2004, 21, 1.
- 20. Blunt, J. W.; Copp, B. R.; Munro, M. H.; Northcote, P. T.; Prinsep, M. R.; Nat. Prod. Rep 2005, 22, 15.
- 21. Blunt, J. W.; Copp, B. R.; Munro, M. H.; Northcote, P. T.; Prinsep, M. R.; Nat. Prod. Rep 2006, 23, 26.
- 22. Blunt, J. W.; Copp, B. R.; Hu, W. P.; Munro, M. H.; Northcote, P. T.; Prinsep, M. R.; Nat. Prod. Rep 2007, 24, 31.
- 23. Blunt, J. W.; Copp, B. R.; Hu, W. P.; Munro, M. H.; Northcote, P. T.; Prinsep, M. R.; Nat. Prod. Rep. 2008, 25, 35.
- 24. Fenical, W.; Jensen, P. R.; Nature Chem. Biol. 2006, 2, 666.
- 25. Bugni, T. S.; Richards, B.; Bhoite, L.; Cimbora, D.; Harper, M. K.; Ireland, C. M.; J. Nat. Prod 2008, 71, 1095.
- 26. Bugni, T. S.; Harper, M. K.; McCulloch, M. W. B.; Reppart, J.; Ireland, C. M.; Molecules 2008, 13, 1372.
- 27. Jensen, P. R.; Mincer, T. J.; William, P. G.; Fenical, W.; Antoine van Leeuwenhoek 2005, 87, 43.
- 28. Haefner, B.; Drug Discov. Today 2003, 8, 536.
- 29. Schwartsmann, G.; Rocha, A. B.; Mattei, J.; Lopes, R. M.; Expert Opin. Investig. Drugs 2003, 12, 1367.
- 30. Newman, D. J.; Cragg, G. M.; J. Nat. Prod. 2004, 67, 1216.
- 31. Class, S.; Chem. Eng. News 2002, 80, 39.
- 32. Fardelone, L. C.; Branchi, B. A.; Rev. FAE 2006, 9, 139.
- 33. Hileman, B.; Chem. Eng. News 2002, 80, 60.
- 34. Hunt, B.; Vincent, A. C.; Ambio 2006, 35, 57.
- 35. Molinski, T. F.; Dalisay, D. S.; Lievens, S. L.; Saludes, J. P.; Nat. Rev. Drug Disc 2009, 8, 69.
-
36http://www.pharmamar.com, acessada em Novembro 2008.
» link -
37http://www.nereuspharm.com, acessada em Dezembro 2008.
» link - 38. Fenical, W.; Jensen, P. R,; Palladino, M. A.; Lam, K. S.; Lloyd, G. K.; Potts, B. C.; Bioorg. Med. Chem. 2009, in press.
- 39. Rouhi, A. M.; Chem. Eng. News 2006, 84, 67.
- 40. Lee, W. W.; Benitez, A.; Goodman, L.; Baker, B. R.; J. Am. Chem. Soc 1960, 82, 2648.
- 41. Roberts, W. K.; Dekker, C. A.; J. Org. Chem. 1967, 32, 816.
- 42. Pomponi, S. A.; Oceanography 2001, 14, 78.
- 43. Suckling, C. J.; Sci. Prog. Edin. 1991, 75, 323.
- 44. Cohen, S. S.; Med. Biol. 1976, 54, 299.
- 45. Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K. W.; Biochemistry 1996, 35, 13303.
- 46. Zewail-Foote, M.; Hurley, L. H. J.; Med. Chem. 1999, 42, 2493.
- 47. Erba, E.; Bergamaschi, D.; Bassano L.; Damia, G.; Ronzoni, S.; Faircloth, G. T.; D'Incalci, M.; Eur. J. Cancer 2001, 37, 97.
- 48. Bonfanti, M.; La Valle, E.; Fernandez Sousa Faro, J. M.; Faircloth, G.; Caretti, G.; Mantovani, R.; D'Incalci, M.; Anti-Cancer Drug Des. 1999, 14, 179.
- 49. Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K. W.; Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 6775.
- 50. Kanzaki, A.; Takebayashi, Y.; Ren, X. Q.; Miyashita, H.; Mori, S.; Akiyama, S.; Pommier, Y.; Mol. Cancer Ther. 2002, 1, 1327.
- 51. Takebayashi, Y.; Pourquier, P.; Zimonjic, D. B.; Nakayama, K.; Emmert, S.; Ueda, T.; Urasaki, Y.; Kanzaki, A.; Akiyama, S.; Popescu, N.; Kraemer, K. H.; Pommier, Y.; Nature Medicine 2001, 7, 961.
- 52. Aune, G. J.; Furuta, T.; Pommier, Y.; Anti-Cancer Drugs 2002, 13, 545.
- 53. Zewail-Foote, M.; Li, V. S.; Kohn, H.; Bearss, D.; Guzman, M.; Hurley, L. H.; Chem. Biol. 2001, 8, 1033.
- 54. Damia, G.; Silvestri, S.; Carrassa, L.; Filiberti, L.; Faircloth, G. T.; Liberi, G.; Foiani, M.; D'Incalci, M.; Int. J. Cancer 2001, 92, 583.
- 55. Puchalski, T. A.; Ryan, D. P.; Garcia-Carbonero, R.; Demetri, G. D.; Butkiewicz, L.; Harmon, D.; Seiden, M. V.; Maki, R. G.; Lopez-Lazaro, L.; Jimeno, J.; Guzman, C.; Supko, J. G.; Cancer Chemother. Pharmacol. 2002, 50, 309.
-
56http://www.emea.epar.eu, acessada em Dezembro 2008.
» link -
57http://www.clinicaltrials.gov, acessada em Novembro 2008.
» link - 58. Martinez, E. J.; Corey, E. J.; Org. Lett. 2000, 2, 993.
- 59. Olivera, B. M.; Gray, W. R.; Zeikus, R.; McIntosh, J. M.; Varga, J.; Rivier, J.; De Santos, V.; Cruz, L. J.; Science 1985, 230, 1338.
- 60. Olivera, B. M.; Cruz, L. J.; De Santos, V.; Lecheminant, G. W.; Griffin, D.; Zeikus, R.; McIntosh, J. M.; Galyean, R.; Varga, J.; Gray, W. R.; Rivier, J.; Biochemistry 1987, 26, 2086.
- 61. Olivera, B. M.; Rivier, J.; Clark, C.; Ramilo, C. A.; Corpuz, G. P.; Abogadie, F. C.; Mena, E. E.; Woodward, S. R.; Hillyard, D. R.; Cruz, L. J.; Science 1990, 249, 257.
- 62. Olivera, B. M.; Ann. Rev. Ecol. Syst. 2002, 33, 25.
- 63. Terlau, H.; Olivera, B. M.; Physiol. Rev 2004, 84, 41.
- 64. Olivera, B. M.; Teichert, R. W.; Mol. Intervent. 2007, 7, 251.
- 65. Berker, S.; Terlau, H.; Appl. Microbiol. Biotechnol. 2008, 79, 1.
- 66. McIntosh, J. M.; Gardner, S.; Luo, S.; Garret, J. E.; Yoshikami, D.; Eur. J. Pahrmacol 2000, 393, 205.
- 67. England, L. J.; Imperial, J.; Jacobsen, R.; Craig, A. G.; Gulyas, J.; Akhtar, M.; Rivier, J.; Julius, D.; Olivera, B. M.; Science 1998, 281,575.
- 68. Möller, C.; Marí, F.; Biochem. J. 2007, 404, 413.
- 69. Ekberg, J.; Craik, D. J.; Adams, D. J.; Int. J. Biochem. Cell Biol. 2008, 40, 2363.
- 70. Bowersox, S. S.; Luther, R.; Toxicon 1998, 36, 1651.
- 71. Prommer, E. E.; Am. J. Hospice Palliat. Med. 2005, 22, 369.
- 72. Williams, J. A.; Day, M.; Heavner, J. E.; Expert Opin. Pharmacother. 2008, 9, 1575.
- 73. Koltz, U.; Int. J. Clin. Pharm. Ther. 2006, 44, 478.
-
74http://www.elan.com, acessada em Novembro 2008.
» link - 75. Proksch, P.; Edrada, R. A.; Ebel, R.; Appl. Microbiol. Biotechnol. 2002, 59, 125.
- 76. Fenical, W.; Trends Biotechnol. 1997, 15, 339.
- 77. Cragg, G. M.; Newman, D. J.; Cancer Invest. 1999, 17, 153.
- 78. Cragg, G. M.; Newman, D. J.; Expert Opin. Investig. Drugs 2000, 9, 2783.
- 79. Sipkema, D.; Osinga, R.; Schatton, W.; Mendola, D.; Tramper, J.; Wijffels, R. H.; Biotechnol. Bioeng. 2005, 90, 201.
- 80. Pomponi, S. A.; Willoughby, R. Em Sponges in time and space; van Soest, R.; van Kempen, T. M. G.; Braekman, J. C., eds.; Balkema: Rotterdam, 1994, p. 395.
- 81. Pomponi, S. A.; Willoughby, R.; Kaighn, M. E., Wright, A. E. Em Invertebrate cell culture: Novel directions and biotechnology applications; Maramorosch, K.; Mitsuhashi, J., eds.; Science Publishers: Enfield, 1997, p. 231.
- 82. Pomponi, S. A.; Willoughby, R.; Wright, A. E.; Pecorella, C.; Sennett, S. H.; Lopez, J.; Samples, G. Em New Developments in Marine Biotechnology; Le Gal, Y.; Halvorson, H. O., eds.; Plenum Press: New York, 1998, p. 73.
- 83. Fenical, W.; Chem. Rev. 1993, 93, 1673.
- 84. Zhang, L.; An, R.; Wang, J.; Sun, N.; Zhang, S.; Hu, J.; Kuai, J.; Curr. Opin. Microbiol. 2005, 8, 276.
- 85. Kelecon A.; An. Acad. Bras. Ciên. 2002, 74, 151.
- 86. Williams, P. G.; TiBtech. 2009, 29, 45.
- 87. Jensen, P. R.; Fenical, W. Em Drugs from the sea; Fusetani, N., ed.; Karger: Tokyo, 2000, p. 6.
- 88. Fenical, W.; Jensen, P. R. Em Natural Products: Drug Discovery and Therapeutic Medicine; Zhang, L.; Demain, A. L., eds.; Humana Press: Totowa, 2005, p. 315.
- 89. Paterson, I.; Anderson, E. A.; Science 2005, 310, 451.
- 90. Wender, P. A.; Verma, V. A.; Paxton, T. J.; Pillow, T. H.; Acc. Chem. Res. 2008, 41, 40.
- 91. Ganesan, A.; Drug Discov. Today 2001, 6, 238.
- 92. Barreiro, E. J.; Rev. Virtual Quim. 2009, 1, 26.
- 93. Rinehart, K. L.; Gloer, J. B.; Cook, J. C.; J. Am. Chem. Soc. 1981, 103, 1857.
- 94. Crampton, S. L.; Adams, E. G.; Kuentzel, S. L.; Li, L. H.; Badiner, G.; Bhuyan, B. K.; Cancer Res. 1984, 44, 1796.
- 95. Vera, M. D.; Joullié, M. M.; Med. Res. Rev. 2002, 22, 102.
- 96. Rinehart, K. L.; Med. Res. Rev. 2000, 20, 1.
- 97. Sakai, R.; Rinehart, K. L.; Kishore, V.; Kundu, B.; Faircloth, G.; Gloer, J. B.; Carney, J. R.; Namikoshi, M.; Sun, F.; Hughes Jr, R. G.; Garcia-Gravalos, D.; de Quesada, T. G.; Wilson, G. R.; Heid, R. M.; J. Med. Chem. 1996, 39, 2819.
- 98. Simmons, T. L.; Adrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W. G.; Mol. Cancer Ther. 2005, 4, 333.
- 99. Urdiales, J. L.; Morata, P.; Nunez De Castro, I.; Sanchez-Jimenez, F.; Cancer Lett 1996, 102, 31.
- 100. Erba, E.; Serafini, M.; Gaipa, G.; Tognon, G.; Marchini, S.; Celli, N.; Rotilio, D.; Broggini, M.; Jimeno, J.; Faircloth, G.; Biondi, A.; D'Incalci, M.; Br. J. Cancer. 2003, 89, 763.
- 101. Broggini, M.; Marchini, S. V.; Galliera, E.; Borsotti, P.; Taraboletti, G.; Erba, E.; Sironi, M.; Jimeno, J.; Faircloth, G. T.; Giavazzi, R.; D'Incalci, M.; Leukemia 2003, 17, 52.
- 102. Jimeno, J.; Faircloth, G.; Sousa-Faro, J. M. F.; Scheuer, P.; Rinehart, K.; Mar. Drugs 2004, 1, 14.
- 103. Gomez, S. G.; Bueren, J. A.; Faircloth, G. T.; Jimeno, J.; Albella, B.; Exp. Hematol. 2003, 31, 1104.
- 104. Hamann, M. T.; Scheuer, P. J.; J. Am. Chem. Soc. 1993, 115, 5825.
- 105. Hamann, M. T.; Otto, C. S.; Scheuer, P. J.; J. Org. Chem. 1996, 61, 6594.
- 106. Singh, R.; Sharma, M.; Joshi, P.; Rawat, D.; Anticancer Agents Med. Chem. 2008, 8, 603.
- 107. Garcıca-Rocha, M.; Bonay, P.; Avila, J.; Cancer Lett. 1996, 99, 43.
- 108. Suárez, Y.; González, L.; Cuadrado, A.; Berciano, M.; Miguel Lafarga, M.; Muñoz, A.; Mol. Cancer Ther. 2003, 2, 863.
- 109. Janmaat, M. L.; Rodriguez, J. A; Jimeno, J.; Kruty, F. A.; Giaccone, G.; Mol. Pharmacol, 2005, 68, 502.
- 110. Manjo, G.; Joris, I.; Am. J. Pathol. 1995, 146, 3.
- 111. Pettit, G. R.; Kamano, Y.; Herald, C. L.; Tuinman, A. A.; Boettner, F. E.; Kizu, H.; Schmidt, J. M.; Baczynkyji, L.; Tomer, K. B.; Bontems, R. J.; J. Am. Chem. Soc. 1987, 109, 6883.
- 112. Pettit, G. R.; Kamano, Y.; Dufresne, C.; Cerny, R. L.; Herald, C. L.; Schmidt, J. M.; J. Org. Chem. 1989, 54, 6005.
- 113. Bai, R.; Pettit, G. R.; Hamel, E.; J. Biol. Chem. 1990, 265, 17141.
- 114. Hamel, E.; Biopolymers 2002, 66, 142.
- 115. Kindler, H. L.; Tothy, P. K.; Wolff, R.; McCormack, R. A.; Abbruzzese, J. L.; Mani, S.; Wade-Oliver, K. T.; Vokes, E. E.; Invest. New Drugs 2005, 23, 489.
- 116. de Jonge, M. J. A.; van der Gaast, A.; Planting, A. S. T.; van Doorn, L.; Lems, A.; Boot, I.; Wanders, J.; Satomi, M.; Verweij, J.; Clin. Cancer Res. 2005, 11, 3806.
- 117. Patel, S.; Keohan, M. L.; Saif, M. W.; Rushing, D.; Baez, L.; Feit, K.; DeJager, R.; Anderson, S.; Cancer 2006, 107, 2881.
- 118. Riely, G. J.; Gadgeel, S.; Rothman, I.; Saidman, B.; Sabbath, K.; Feit, K.; Kris, M. G.; Rizvi, N. A.; Lung Cancer 2007, 55, 181.
- 119. Mross, K. H. K.; Herbst, K.; Berdel, W. E.; Korfel, A.; von Broen, I.-M.; Bankmann, Y.; Hossfeld, D. K.; Onkologie 1996, 19, 405.
- 120. Wolff, I.; Bruntsch, U.; Cavalli, F.; Proc. Am. Soc. Clin. Oncol. 1997, 16, A783 (abstr).
- 121. Allen, S. W.; Villalona-Calero, M. A.; Jakimowicz, K.; Fram, R.; O'Mara, V.; Kolitz, J. E.; Gallagher, M. A.; van Echo, D.; Fischkoff, S.; O'Dwyer, P.; Proc. Am. Assoc. Cancer Res. 1997, 388, A1498 (abstr).
- 122. Villalona-Calero, M. A.; Baker, S. D.; Hammond, L.; Aylesworth, C.; Eckhardt, S. G.; Kraynak, M.; Fram, R.; Fischkoff, S.; Velagapudi, R.; Toppmeyer, D.; Razvillas, B.; Jakimowicz, K.; von Hoff, D. D.; Rowinsky, E.; J. Clin. Oncol. 1998, 14, 2770.
- 123. Mross, K.; Berdel, W. E.; Fiebig, H. H.; Velagapudi, R.; von Broen, I.-M.; Bankmann, Y.; Hossfeld, D. K.; Ann. Oncol. 1998, 9, 1323.
- 124. Supko, J. G.; Lynch, T. J.; Clark, J. W.; Fram, R.; Allen, L. F.; Velagapudi, R.; Kufe, D. W.; Ede, J. P., Jr.; Cancer Chemother. Pharmacol. 2000, 46, 319.
- 125. Smyth, J.; Boneterre, M. E.; Schellens, J. H. M.; Calvert, H.; Greim, G.; Wanders, J.; Hanauske, A.; Ann. Oncol. 2001, 12, 509.
- 126. Kerbrat, P.; Dieras, V.; Pavlidis, N.; Ravaud, A.; Wanders, J.; Fumoleau, P.; Eur. J. Cancer 2003, 39, 317.
- 127. Marks, R. S.; Graham, D. L.; Sloan, J. A.; Hillman, S.; Fishkoff, S.; Krook, J. E.; Okuno, S. H.; Mailliard, J. A.; Fitch, T. R.; Addo, F.; Am. J. Clin. Oncol. 2003, 26, 336.
- 128. Ebbinghaus, S.; Hersh, E.; Cunningham, C. C.; O' Day, S.; McDermott, D.; Stephenson, J.; Richards, D. A.; Eckardt, J.; Haider, O. L.; Hammond, L. A.; J. CLin. Oncol. 2004, 22, 7530.
- 129. Mita, A. C.; Hammond, L. A.; Garrison, M.; McCreery, H.; Weiss, G.; Schwartz, G. H.; Jekunen, A.; van Dijk, S.; Rowinsky, E. K.; Proc. Am. Soc. Clin. Oncol. 2003, 22, 906.
- 130. Bhakuni, D. S.; Rawat D. S. Em Bioactive Marine Natural Products; Bhakuni, D. S.; Rawat D. S., eds.; Anamaya Publishers: New Delhi, 2006, cap. 10.
-
131http://findarticles.com/p/articles/mi_m0EIN/is_/ai_108611542, acessada em Dezembro 2008.
» link - 132. Shioiri, T.; Hayashi, K.; Hamada, Y.; Tetrahedron 2007, 63, 1913.
- 133. Mordant, C.; Reymond, S.; Tone, H.; Lavergne, D.; Touati, R.; Ben Hassine, B.; Ratovelomanana-Vidal, V.; Genet, J.-P.; Tetrahedron 2007, 63, 6115.
- 134. Cella, R.; Venturoso, R. C.; Stefani, H. A.; Tetrahedron Lett. 2008, 49, 16.
- 135. Pettit, G. R.; Day, J. F.; Hartwell, J. L.; Wood, H. B.; Nature 1970, 227, 962.
- 136. Pettit, G. R.; Herald, C. L.; Doubek, D. L.; Herald, D. L.; Arnold, E.; Clardy, J.; J. Am. Chem. Soc. 1982, 104, 6846.
- 137. AlKatib, A. M.; Smith, M. R.; Kamanda, W. S.; Pettit, G. R.; Hamdan, M.; Mohamed, A. N.; Chelladurai, B.; Mohammad, R. M.; Clin. Cancer Res. 1998, 4, 1305.
- 138. Farrow, B.; Thomas, R. P.; Wang, X. F.; Evers, B. M.; Int. J. Gastrointest. Cancer 2002, 32, 63.
- 139. Dowlati, A.; Lazarus, H. M.; Hartman, P.; Jacobberger, J. W.; Whitacre, C.; Gerson, S. L.; Ksenich, P.; Cooper, B. W.; Gottlieb, M.; Murgo, A. J.; Clin. Cancer Res. 2003, 9, 5929.
- 140. Pettit, G. R.; Herald, C. L.; Hogan, F. Em Anticancer Drug Development; Baguley, B. C.; Kerr, D. J., eds.; Academic Press: San Diego, 2002, cap. 12.
- 141. Clamp, A.; Jayson, G. C.; Anti-Cancer Drugs 2002, 13, 673.
- 142. Varterasian, M. L.; Mohammad, R. M.; Eilender, D. S.; Hulburd, K.; Rodriguez, D. H.; Pemberton, P. A.; Pluda, J. M.; Dan, M. D.; Pettit, G. R.; Chen, B. D.; Al-Katib, A. M.; J. Clin. Oncol. 1998, 16, 56.
- 143. Clamp, A. R.; Blackhall, F. H.; Vasey, P.; Soukop, M.; Coleman, R.; Halbert, G.; Robson, L.; Jayson G. C.; Brit J. Can. 2003, 89, 1152.
- 144. Newman, D. J. Em Antitumor Agents from Natural Sources; Kingston, D. G. I.; Cragg, G. M.; Newman, D. J., eds.; CRC Press: Boca Raton, 2004.
- 145. Mendola, D.; Biomol. Eng 2003, 20, 441.
- 146. Davidson, S. K.; Allen, S. W.; Lim, G. E.; Anderson, C. M.; Haygood, M. G.; Appl. Environ. Microbiol. 2001, 67, 4531.
- 147. Davidson, S. K.; Haygood, M. G.; Biol. Bull. 1999, 196, 273.
- 148. Trost, B. M.; Dong, G.; Nature 2008, 456, 485.
- 149. Kageyama, M.; Tamura, T.; Nantz, M. H.; Roberts, J. C.; Somfai, P.; Whritenour, D. C.; Masamune, S.; J. Am. Chem. Soc. 1990, 112, 7407.
- 150. Evans, D. A.; Carter, P. H.; Carreira, E. M.; Charette, A. B.; Prunet, J. A.; Lautens M.; J. Am. Chem. Soc. 1999, 121, 7540.
- 151. Ohmori, K.; Ogawa, Y.; Obitsu, T.; Ishikawa, Y.; Nishiyama, S.; Yamamura, S.; Angew. Chem., Int. Ed. 2000, 39, 2290.
- 152. Wender, P. A.; Baryza, J. L.; Brenner, S. E.; Clarke, M. O.; Craske, M. L.; Horan, J. C.; Meyer, T.; Curr. Drug Discov. Technol. 2004, 1, 1.
- 153. Hung, D. T.; Nerenberg, J. B.; Schreiber, S. L.; J. Am. Chem. Soc. 1996, 118, 11054.
- 154. Harried, S. S.; Yang, G.; Strawn, M. A.; Myles, D. C.; J. Org. Chem. 1997, 62, 6098.
- 155. Marshall, J. A.; Johns, B. A.; J. Org. Chem. 1998, 63, 7885.
- 156. Paterson, I.; Florence, G. J.; Gerlach, K.; Scott, J. P.; Angew. Chem., Int. Ed. 2000, 39, 377.
- 157. Paterson, I.; Florence, G. J.; Eur. J. Org. Chem. 2003, 12, 2193.
- 158. Smith III, A. B.; Freeze, B. S.; Xian, M.; Hirose, T.; Org. Lett. 2005, 7, 1825.
- 159. Gunasekera, S. P.; Gunasekera, M.; Longley, R. E.; Schulte, G. K.; J. Org. Chem. 1990, 55, 4912.
- 160. Burlingame, M. A.; Mendoza, E.; Ashley, G. W.; Myles, D. C.; Tetrahedron Lett. 2006, 47, 1209.
- 161. Smith III, A. B.; Freeze, B. S.; Tetrahedron 2008, 64, 261.
- 162. Rosenkranz, H. S.; Day, B. W.; Biochemistry 1996, 35, 243.
- 163. Myles, D. C.; Ann. Rep. Med. Chem. 2002, 37, 125.
- 164. ter Harr, E.; Kowalski, R. J.; Hammel, E.; Lin, C. M.; Longley, R. E.; Gunasekera, S. P.; Buey, R. M.; Barasoain, I.; Jackson, E.; Meyer, A.; Giannakakou, P.; Paterson, I.; Mooberry, S.; Andreu, J. M.; Diaz, J. F.; Chem. Biol. 2005, 12, 1269.
- 165. Jordan, M. A.; Kamath, K.; Manna, T.; Okouneva, T.; Miller, H. P.; Davis, C.; Littlefield, B. A.; Wilson, L.; Mol. Cancer Ther. 2005, 4, 1086.
- 166. Newman, S.; Curr. Opin. Invest. Drugs 2007, 8, 1057.
- 167. Atieh, D. M.; Vahdat, L. T.; Breast Diseases 2007, 18, 21.
-
168http://www.marinebiotech.org/spisulosine.html, acessada em Dezembro 2008.
» link - 169. Cuadros, R.; Montejo de Garcini, E.; Wandosell, F.; Faircloth, G.; Fernandez-Sousa, J. M.; Avila, J.; Cancer Lett 2000, 152, 23.
- 170. Salcedo, M.; Cuevas, C.; Alonso, J. L.; Otero, G.; Faircloth, G.; Fernandez-Sousa, J. M.; Avila, J.; Wandosell, F.; Apoptosis 2007, 12, 395.
- 171. Natori, T.; Koezuka, Y.; Higa, T.; Tetrahedron Lett. 1993, 34, 5591.
- 172. Morita, M.; Motoki, K.; Akimoto, K.; Natori, T.; Sakai, T.; Sawa, E.; Yamaji, K.; Koezuka, Y.; Kobayashi, E.; Fukushima, H.; J. Med. Chem. 1995, 38, 2176.
- 173. Franck, R. W.; Tsuji, M.; Acc. Chem. Res. 2006, 39, 692.
- 174. Hayakawa, Y.; Rovero, S.; Forni, G.; Smyth, M. J.; Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9464.
- 175. Natori, T.; Morita, M.; Akimoto, K.; Koezuka, Y.; Tetrahedron 1994, 50, 2771.
- 176. Elices, M.; Grant, W.; Harper, C.; Guillen, M. J.; Cuevas, C.; Faircloth, G.; Aviles, P.; Proc. Am. Assoc. Cancer Res. 2005, 46, Abs. 623.
- 177. Greiner, T.; Maier, A.; Bausch, N.; Fiebig, H.; LePage, D.; Guillen, M. J.; Cuevas, C.; Aviles, P.; AACR 2007, Abs. C60.
- 178. LePage, D.; Sasak, H.; Guillen, M. J.; Grant, W.; Cuevas, C.; Aviles, P.; AACR 2007, Abs. C62.
- 179. LePage D.; Sasak, H.; Cheney, L.; Oyama, S.; Cuevas, C.; Aviles, P.; Maxuitenko, Y.; Faircloth, G.; AACR 2007, Abs. 1519.
- 180. Ocio, E. M.; Maiso, P.; Chen, X.; Garayoa, M.; Alvarez-Fernandez, S.; San-Segundo, L.; Vilanova, D.; Lopez-Corral, L.; Montero, J. C.; Hernandez-Iglesias, T.; de Alava, E.; Galmarini, C.; Aviles, P.; Cuevas, C.; San-Miguel, J. F.; Pandiella A.; Blood 2008, DOI 10.1182.
- 181. Elices, M.; Sasak, H.; Caylor, T.; Grant, W.; Guillen, M. J.; Martin, J.; Cuevas, C.; Aviles, P.; Faircloth, G.; Proc. Am. Assoc. Cancer Res. 2005, 46, Abs. 5882.
- 182. Sorbera, L. A.; Castañer, R. M.; Leeson, P. A.; Drugs Fut. 2000, 25, 551.
- 183. Gingras, D.; Boivin, D.; Deckers, C.; Gendron, S.; Barthomeuf, C.; Beliveau, R.; Anti-Cancer Drugs 2003 14, 91.
- 184. Feling, R. H.; Buchanan, G. O.; Mincer, T. J.; Kauffman, C. A.; Jensen, P. R.; Fenical, W.; Angew. Chem., Int. Ed. 2003, 42, 355.
- 185. Richarson, P. G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S. V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; Seigel, D.; Orlowski, R. Z.; Kuter, D.; Limentani, S. A.; Lee, S.; Hideshima, T.; Esseltine, D. L.; Kauffman, M.; Adams, J.; Schenkein, D. P.; Anderson, K. C.; N. Engl. J. Med. 2003, 348, 2609.
- 186. Adams, J.; Proteasome Inhibitors in Cancer Therapy, Humana Press: Totowa-New Jersey, 2004.
- 187. Reddy, L. R.; Saravanan, P.; Corey, E. J.; J. Am. Chem. Soc. 2004, 126, 6230.
- 188. Endo, A.; Danishefsky, S. J.; J. Am. Chem. Soc. 2005, 127, 8298.
- 189. Nicholson, B.; Lloyd, G. K.; Miller, B. R.; Palladino, M. A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S. T. C.; Anti-Cancer Drugs 2006, 17, 25.
Datas de Publicação
-
Publicação nesta coleção
22 Maio 2009 -
Data do Fascículo
2009
Histórico
-
Aceito
11 Mar 2009 -
Recebido
15 Jan 2009