Resumo
A view of the general aspects involving the 2D NMR spectroscopy using inverse detection and field gradient techniques is presented through the analysis of a sesquiterpene.
2D NMR; inverse detection; field gradient
2D NMR; inverse detection; field gradient
DIVULGAÇÃO
RMN 2D: detecção inversa e gradiente de campo na determinação estrutural de compostos orgânicos
Carlos R. Kaiser
Instituto de Química - Universidade Federal do Rio de Janeiro - Fundão - CT - Bloco A - 21949-900 - Rio de Janeiro - RJ
Recebido em 13/7/98; aceito em 5/7/99
2D NMR: inverse detection and field gradient in structure determination of organic compounds. A view of the general aspects involving the 2D NMR spectroscopy using inverse detection and field gradient techniques is presented through the analysis of a sesquiterpene.
Keywords: 2D NMR; inverse detection; field gradient.
RESSONÂNCIA MAGNÉTICA NUCLEAR
A Ressonância Magnética Nuclear (RMN) é sem dúvida uma das mais poderosas técnicas espectroscópicas no estudo de aspectos estruturais e dinâmicos de moléculas orgânicas em solução. Tanto que na atualidade é muito comum fazer medidas de RMN para uma determinada substância, mesmo antes de se obter um espectro de infravermelho ou um espectro de massa. Esta constatação está diretamente relacionada com a evolução técnica dos equipamentos de RMN nesta última década. Por exemplo: magnetos supercondutores com maior estabilidade do campo magnético e versatilidade no ajuste da homogeneidade; hardwares que permitem eficiente elaboração e precisão na programação das sequências de pulsos, de forma a acomodar as mais variadas técnicas multidimensionais (2D, 3D, etc.); softwares que fornecem uma interface mais simples e rápida com o hardware, facilitando o controle do instrumento. As melhorias nos equipamentos geralmente vem acompanhadas da introdução de novas sequências de pulsos. Todavia, apesar de sempre serem instigantes para o químico espectroscopista, novas técnicas de pulsos levam um longo tempo até que sejam plenamente absorvidas pela grande maioria dos usuários1-5.
Ainda é rotina obter espectros em uma dimensão (1D) de 1H, 13C{1H} e DEPT de um determinado composto, mesmo que em muitas situações estas duas últimas técnicas exijam um tempo de medida muito grande devido a baixa sensibilidade inerente do núcleo de 13C (1.59% relativa ao 1H) e normalmente, a pouca quantidade de amostra disponível (< 0.03 mg/mol). À parte das possíveis dificuldades experimentais, está comprovado, através dos anos, que os dados de deslocamentos químicos de 13C são importantes ferramentas em uma determinação estrutural2,3,5-7.No entanto, também ficou demonstrado que a metodologia de comparar os dados obtidos com os dados de compostos análogos descritos na literatura merece sempre cautela. Isto porque vários destes registros literários sofrem de imprecisões devido as limitações dos equipamentos e as dificuldades de assinalamento para moléculas estruturalmente complexas.
RMN EM DUAS DIMENSÕES
Não tão difundida quanto obter espectros 1D de compostos orgânicos, é a prática de obter espectros em duas dimensões (2D), homo (1H,1H) e heteronucleares (1H,13C)1-7. As técnicas 2D permitem correlacionar hidrogênios e/ou carbonos que possuam alguma relação escalar (nJH,H, nJC,H,nJC,C )6,7,9 ou espacial (H,H- ou H,C-NOE)2,3,6,8 entre si. Desta forma é possível fazer um mapeamento estrutural mais preciso do que utilizando somente as técnicas 1D e dados de possíveis compostos similares da literatura, principalmente na análise de estruturas mais complexas. Não obstante, é sempre recomendável que o usuário de RMN 2D esteja familiarizado com a influência de fatores como eletronegatividade ou anisotropia magnética ou compressão estérica sobre os deslocamentos químicos e/ou constantes de acoplamentos6,7,10.
Um resumo das técnicas 2D mais difundidas mostra-se então bastante oportuno:
COSY (homonuclear COrrelation SpectroscopY) - com esta técnica pode-se estabelecer as correlacões entre os hidrogênios que estão acoplados por 2-3JH,H (acoplamentos geminais e vicinais, mensuráveis no espectro 1D) e assim discernir a multiplicidade dos sinais observados no espectro de RMN1H. Eventualmente, sinais devidos a 4-6JH,H são também visíveis (acoplamentos a longa distância, raramente mensuráveis no espectro 1D)1-7,9.
COSYLR (COSY for Long Range couplings) - uma variante do COSY, mas neste caso, evidenciando as correlações devido a acoplamentos à longas distâncias. Em geral, as correlações devidas a acoplamentos 2-3JH,H também estão presentes no espectro, mas consideravelmente minimizadas1,5,9.
DQF-COSY (Double Quantum Filter-COSY) - outra variante do COSY, sendo que neste caso os sinais da diagonal estão ausentes ou, no mínimo, bastante minimizados. É muito útil para observar correlações de hidrogênios com dH muito próximos uns dos outros. Outro fator positivo é o de que muitas correlações devido a acoplamentos à longas distâncias 4-6JH,H ficam visíveis1-3,5,7,9.
TOCSY (TOtal Correlation SpectroscopY) - tem o aspecto de um COSY, mas as correlações envolvem a condição HOmonuclear HArtmann-Hahn (HOHAHA). Em uma visão simplificada, revela correlações entre hidrogênios não necessariamente acoplados, mas que possuem um hidrogênio vizinho comum ao qual ambos estão acoplados. É uma técnica muito útil na análise de compostos que possuem um carbono sem hidrogênios ou um heteroátomo, separando dois subsistemas na mesma molécula1,5.
NOESY (Nuclear Overhauser Enhancement SpectroscopY) - também tem o aspecto de um COSY, contudo as correlações envolvem interações devidas ao NOE entre os hidrogênios que estão espacialmente próximos (em geral, menor do que 4 Å). Através desta técnica é possível estabelecer a configuração relativa de cada hidrogênio na molécula e geralmente a geometria molecular como um todo. A mesma informação pode ser obtida da difração de Raio-X, mas apenas para compostos na forma de um sólido cristalino1-5,7.
HETCOR (HETeronuclear CORrelation spectroscopy) - com este experimento pode-se assinalar os carbonos que contém hidrogênios, caso já tenham sido assinalados todos os hidrogênios do espectro de RMN1H, ou vice e versa, pois a técnica depende dos acoplamentos 1JC,H. O espectro é apresentado com o eixo horizontal (dimensão F2) correspondendo aos dC e o eixo vertical (dimensão F1) correspondendo aos dH1-3,5-7.
COLOC (COrrelation spectroscopy for LOng-range Couplings) - o espectro é apresentado como um HETCOR contudo as correlações referem-se aos acoplamentos nJC,H (n > 2). Infelizmente os sinais devidos aos 1 JC,H estão invariavelmente presentes, o que torna imprescindível um experimento HETCOR prévio. As informações obtidas das correlações nJC,H são muito importantes em uma determinação estrutural, principalmente para estabelecer a posição de carbonos não hidrogenados1-3,5.
Apesar das vantagens em termos de informações estruturais que as técnicas 2D tradicionais oferecem, o tempo necessário para executar certos experimentos, principalmente os heteronucleares, sempre foi uma grande limitação. Por exemplo, para obter um espectro HETCOR e um COLOC em um equipamento de 7.05 Tesla (300 MHz para o 1H), de uma amostra de 10 mg com massa molecular ~500u, o pesquisador necessitaria dispor de, no mínimo, 48h/máquina !
RMN 2D COM DETECÇÃO INVERSA
A dificuldade básica em obter os espectros HETCOR e COLOC, principalmente para amostras diluídas, como também ocorre para um experimento 1D de 13C{1H}, reside no fato de que a detecção é feita por intermédio do heteronúcleo pouco sensível (detecção direta no canal de 13C, 15N, etc.). Visando contornar esta limitação, foram criadas técnicas de pulso 2D heteronucleares em que a detecção é feita por intermédio de um núcleo muito mais sensível (detecção indireta no canal de 1H ou 19F ou 31P). Sequências de pulsos engenhosas envolvendo transições multiplo-quânticas, denominadas de HMQC (Heteronuclear MultipleQuantum Coherence) e HMBC (Heteronuclear MultipleBond Coherence) substituíram então os recíprocos HETCOR e COLOC. Os espectros HMQC e HMBC são editados com o eixo horizontal (dimensão F2) correspondendo aos dH e o eixo vertical (dimensão F1) correspondendo aos dC1,5.Não é preciso muito esforço para entender a vantagem da detecção indireta sobre a direta, é apenas uma questão de sensibilidade!
Para otimizar ainda mais este efeito da detecção indireta foram criadas "sondas inversas". As sondas são acessórios permutáveis, que inseridos no vão central dos magnetos, são responsáveis pela emissão da radiofrequência de excitação bem como da detecção do sinal devido a perturbação da amostra. As sondas inversas são construídas de forma que a bobina do canal de 1H fique posicionada como a mais interna, mais próxima do tubo contendo a amostra, em relação a outras bobinas (ex. 13C e 2H), tornando mais sensível a detecção para este núcleo. Tecnicamente, a diferença entre uma sonda direta (bobina de 13C mais interna) e uma inversa, são apenas alguns mi-límetros mas, em termos práticos, consegue-se uma economia de várias horas nas medidas 2D homo e heteronucleares1,5.
RMN 2D COM GRADIENTE DE CAMPO
As sondas inversas podem ainda ser providas de bobinas adicionais de gradiente de campo, tornando possível reduzir ou eliminar os vários ciclos de modulação de fase (phase cycling) no transmissor e no receptor de frequências. Esta modulação de fase faz-se necessária para a perfeita coerência de ordem quântica e cancelamento das ordens não desejadas, nos experimentos 2D citados acima. O gradiente de campo conduz a uma seleção quântica muito mais eficiente e em um tempo muito menor. O espectro gs-2D (gradient selected-2D) quase não possui ruídos (artefatos), que são causados justamente pelo cancelamento imperfeito das ordens quânticas não desejadas usando-se o phase cycling.1-5 As técnicas homonucleares como COSY1,5,11, COSYLR1,5,11,12, DQF-COSY (ex. Figura 1)1,5,13 e TOCSY1,5,14, bem como as técnicas heteronucleares HMQC1,5,15 e HMBC (ex. Figura 4)1,5,15, podem todas ser executadas com gradiente de campo. Além disso várias técnicas muito interessantes foram criadas, principalmente as heteronucleares que fundem duas ou mais técnicas 1D/2D num só experimento 2D:
HSQC-ED (Heteronuclear Single Quantum Coherence - Editing) - tem o aspecto de um HMQC, só que neste caso envolvendo transições quânticas simples e, além disso, é editado com os cruzamentos devidos a grupos CH2 em fase oposta a grupos CH e CH3 como num DEPT135 (ex. Figura 2)1,5,16-20.
HSQC-TOCSY - mostra correlações C,H,H ou seja tem o aspecto de um HSQC mas além das correlações C,H há também correlações H,H como no TOCSY (ex. Figura 3)1,5,15,16,21.
Desde o início da presente década os equipamentos de RMN comerciais podem ser providos de sondas inversas e, mais recentemente, do sistema de gradiente de campo. São acessórios opcionais, contudo, o ganho em tempo de análise e variedade de informações que se pode obter justifica o investimento. São atualmente imprescindíveis na área de Bioquímica (determinação estrutural de proteínas e carboidratos)1,4,5 e já vem se tornando indispensáveis também nas demais áreas da Química. Para ilustrar melhor como algumas das técnicas de RMN 2D envolvendo detecção inversa e gradiente de campo podem ser utilizadas, apresentamos uma análise estrutural de um composto orgânico modelo, o johnstonol22.Os vários aspectos experimentais estão descritos no item Detalhes Técnicos.
APLICAÇÃO NA DETERMINAÇÃO ESTRUTURAL
A fórmula molecular C15H21O3Br2 Cl (442.9546u) do johnstonol foi determinada por espectrometria de massa de alta resolução (EMAR). Já a espectroscopia de infravermelho (IV) revelou a presença de grupamento hidroxila e a ausência de insaturações (C=C ou CºC ou C=O). O johnstonol é um produto natural extraído do molusco marinho Aplysia dactylomela, encontrado inclusive em águas brasileiras, rico em sesquiterpenos com esqueleto chamigreno11,22,23. Estes sesquiterpenos são compostos que apresentam estruturas cíclicas, bicíclicas, etc., contendo halogênios (bromo, cloro) e eventualmente também insaturações, hidroxilas e epóxidos. Até aqui os dados deixam claro que o johnstonol é formado por quatro ciclos saturados, considerando o grau de insaturação da fórmula C15H21O3Br2 Cl e a inexistência de anéis aromáticos, carbonilas, ligações duplas ou triplas.
As medidas de RMN foram feitas com uma amostra de johnstonol a 0.016 mg/mol em 0.6 ml de CDCl3. Do espectro de RMN1H foram extraídos todos os dH e 2-3JH,H , sendo que este mostra (veja projeção na dimensão F2 das Figuras 1-4) quatro singletos entre d 1.10-1.80 (desprezando-se o sinal em d 1.55 devido a água do solvente e o pequeno sinal em d 1.20 devido a alguma impureza) cada um destes com área proporcional para três hidrogênios, sugerindo quatro metilas ligadas a carbonos não hidrogenados; multipletos entre d 2.00-2.20 com área total igual a três; e seis sinais de área igual a um: singleto em d 3.15, dois dubletos em d 3.98 e 2.61 e três duplo dubletos em d 4.60, 4.24 e 2.50.
Através do espectro gs-DQF-COSY (gs = gradient selected) da Figura 1 podemos estabelecer a origem dos acoplamentos escalares observados nos sinais do espectro de RMN1H. Para isto basta correlacionar os sinais observados no espectro 2D entre hidrogênios com dH distintos (veja exemplo na Figura 1A). Com a expansão apresentada na Figura 1B podemos resolver os multipletos entre d 2.00-2.20 (dimensão F2) como sendo constituídos de três duplo dubletos com os dois das extremidades desdobrados de 14.47 e 13.62 Hz (d 2.18, H-4ax) e 14.52 e 12.97 Hz (d 2.07, H-1ax) e o central de 14.52 e 2.82 Hz (d 2.16, H-1eq). O gs-DQF-COSY apresenta ainda três sinais que envolvem acoplamentos a longas distâncias, mas cujas magnitudes não são mensuráveis no espectro de RMN1H : um sinal, bastante intenso, entre os hidrogênios metílicos mais protegidos em d 1.12 (H-13) e d 1.23 (H-14) mostrando serem metilas ligadas ao mesmo carbono e acopladas por 4JH,H ; e dois sinais, de baixa intensidade, dos hidrogênios metílicos H-15 (d 1.40) com os hidrogênios metínicos em d 3.15 (H-10) e d 3.98 (H-9) a quatro (4JH,H) e cinco ligações ( 5JH,H), respectivamente7,9.
O experimento HSQC-ED apresentado na Figura 2 revela que temos dois carbonos metilênicos (CH2), com dois grupos de sinais para cada carbono pois os hidrogênios correspondentes não são equivalentes; quatro carbonos metílicos (CH3); e quatro carbonos metínicos (CH). O HSQC-ED (resumido no Quadro1) revela também que o sinal em d 2.61 não está ligado a um carbono, pois não há sinal no 2D e, assim, trata-se de um hidrogênio hidroxílico que devido à troca química lenta tem um acoplamento de 2.31 Hz (3JH,H) com o hidrogênio carbinólico H-9 em d 3.98 (dC 74.61). Já os deslocamentos químicos dC 59.75 (C-10) e dH 3.15 (H-10) são característicos de um grupo metínico que faz parte de um anel epóxido. No espectro de RMN1H o sinal em dH 3.15 apresenta-se como um singleto, o que indica que este hidrogênio é o único ligado diretamente a um dos dois carbonos deste ciclo de três membros, considerando-se os padrões de acoplamentos 2-3JH,H possíveis6,7.
O HSQC-TOCSY apresentado na Figura 3 mostra, em um só experimento, o que foi observado nos experimentos gs-DQFCOSY e HSQC-ED separadamente. Levando-se em consideração as relações C,H,H representadas no espectro e os deslocamentos químicos envolvidos, podemos gerar as substruturas a e b que são necessariamente delimitadas por carbonos não hidrogenados, como ilustra o Esquema 1. Seguindo o mesmo raciocínio, com as linhas traçadas na Figura 3, podemos isolar outras duas subestruturas, c e d, com ambos os hidrogênios metínicos d 4.60 (H-5) e 4.24 (H-2) em posição axial levando em conta as constantes de acoplamento de 13.62 e 12.97 Hz, respectivamente.

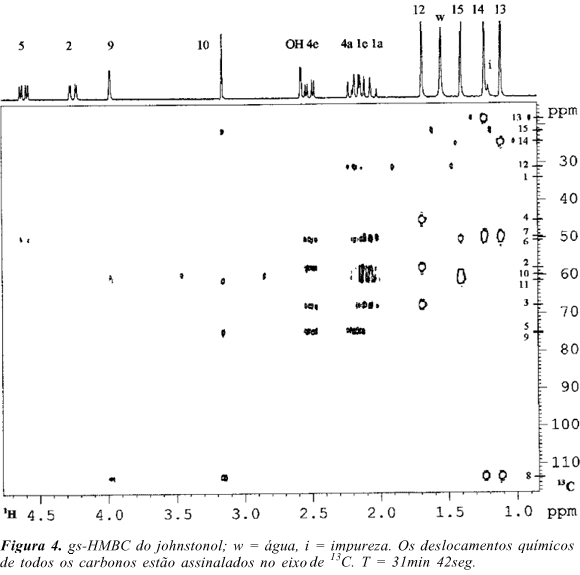
Determinadas as subestruturas deste sesquiterpeno, resta agora definir como estas estão interligadas através dos carbonos não hidrogenados. Para isso podemos utilizar o espectro gs-HMBC da Figura 4. Este experimento envolve os acoplamentos nJC,H (com n ³ 2) que são dependentes das distâncias e dos ângulos de ligação entre os núcleos envolvidos.6 Em geral, as intensidades relativas dos sinais no espectro podem ser diretamente relacionadas com as magnitudes das constantes de acoplamento1,5,24. Como são necessárias várias ampliações para uma melhor visualização do gs-HMBC, utilizamos o Quadro1 para facilitar a análise dos sinais observados no espectro. Partindo-se do subsistema a observa-se que as metilas mais protegidos H-13 e H-14 (dH 1.12 e 1.23 ) mostram quatro correlações cada: uma entre eles (dC 24.60, C-14, e dC 18.19, C-13, respectivamente) e as outras com os carbonos não hidrogenados em dC 49.36, 50.43 e 113.40. Destes três, o primeiro corresponde ao carbono ao qual C-13 e C-14 estão ligados, ou seja C-7 pois este carbono só apresenta correlações com H-13 e H-14. Acontece que os hidrogênios metílicos em dH 1.40 (H-15), do subsistema b, também mostram correlação com o carbono dC 50.43 (C-6), bem como os hidrogênios em dH 3.15 (H-10) e 3.98 (H-9) com o carbono em dC 113.40 (C-8). Consequentemente, podemos fundir a com b em um anel de seis membros onde, de um lado temos o carbono não hidrogenado mais desprotegido e do outro o mais protegido. O carbono C-6 (dC 50.43) também mostra correlações com os hidrogênios metilênicos H-1 (dH 2.07 e 2.16) e H-4 (dH 2.18 e 2.50), sendo portanto um dos pontos de fechamento de outro ciclo. O outro ponto compreende o carbono não hidrogenado C-3 em dC 67.88, pois os hidrogênios metílicos em dH 1.68 (H-12) mostram correlação com C-3 bem como com os carbonos C-2 e C-4 em dC 57.91 (c) e 45.16 (d). O último ciclo envolve o átomo de oxigênio que está ligado aos dois carbonos mais desprotegidos.
A estrutura molecular está definida e ainda com boas indicações da estereoquímica oriunda da dependência angular do acoplamentos nJH,H e nJC,H com n > 1. Pode-se estabelecer a estereoquímica relativa através das interações espaciais H,H observáveis em um experimento NOESY (não é realizado com gradiente de campo pois não conduz a uma boa seleção quântica). Neste ponto, é altamente recomendável utilizar modelos moleculares para visualizar as proximidades espaciais. Em resumo, o experimento revelou que a metila mais protegida H-13 (dH 1.12) localiza-se do mesmo lado do ciclo de seis membros que contém a hidroxíla, pois há apenas correlações com H-15 (dH 1.40), O-H (dH 2.61) e H-10 (dH 3.15). Isto indica também que o hidrogênio H-10 ligado ao anel epóxido está em posição pseudo-axial e que o hidrogênio carbinólico H-9 (dH 3.98) está em posição pseudo-equatorial, justificando o primeiro como singleto pois o ângulo diedro não é favorável ao acoplamento 3JH,H entre ambos (f ~ 90o). A metila H-14 (dH 1.23) está do mesmo lado do ciclo tetraidrofurano, pois há correlações apenas com H-1ax e H-4ax em dH 2.07 e 2.18. A inexistência de correlações da metila H-12 (dH 1.68) com H-14, H-2ax e H-5ax indica que ela está em posição equatorial em um anel com conformação bote. Visto que as magnitudes de 3JHax,Hax e 3JHax,Heq do sistema C-1/C-2 são consideravelmente menores do que para o sistema C-4/C-5, há um ligeira distorção da conformação bote ao redor da ligação C1-C2, de forma que a ligação C2-Br não fique eclipsada com a ligação C3-C12.
A Tabela 1 resume os dados de 1H e 13C referentes ao johnstonol. Vale ressaltar que todos os dados de dC podem ser extraídos dos experimentos 2D heteronucleares, conforme foi exemplificado nas Figuras 2-4.
CONSIDERAÇÕES FINAIS
O propósito deste artigo não foi fazer uma discussão exaustiva da estrutura do johnstonol. A determinação estrutural deste produto natural foi apenas uma maneira encontrada para ilustrar, de forma prática, aspectos gerais e principalmente os avanços técnicos que aconteceram na área de RMN 2D nesta última década.
O reduzido tempo de análise dos experimentos 2D e a qualidade dos espectros são, sem sombra de dúvida, as maiores virtudes das técnicas inversas com gradiente de campo. Assim foi para o exemplo do johnstonol, que consumiu um total de "1h35min" para o gs-DQF-COSY, HSQC-ED, HSQC-TOCSY e gs-HMBC, eliminando ainda a necessidade de um espectro de carbono-13 e um de DEPT135 em 1D (para a presente amostra necessita-se respectivamente de 1h30min e 30min, para obter uma razoável relação sinal/ruído nos espectros). Evidentemente não são todos os casos em que se pode abdicar de um espectro de RMN de 13C. No entanto, são muitos os casos em que toda uma determinação estrutural e assinalamento dos sinais de hidrogênios e carbonos podem ser conduzidos fazendo-se uso apenas de algumas técnicas gs-2D. É uma opção importante para laboratórios de RMN que possuem condições de executar técnicas inversas com gradiente de campo pois, em geral, há mais análises do que equipamentos disponíveis para executá-las.
As técnicas inversas com gradiente de campo deixam ainda aberto um grande campo para o desenvolvimento de novas sequências de pulsos, que resultem em experimentos tão interessantes como o HSQC-ED e o HSQC-TOCSY aqui apresentados. Por exemplo, mesmo fora do presente contexto, vale ressaltar o WATERGATE (WATER supression by GrAdient-Tailored Excitation)25,26 como uma eficiente técnica de supressão do sinal devido a água, principalmente quando usada como solvente em amostras de polissacarídeos e proteínas1,3,5 e o DOSY (Diffusion-Ordered SpectroscopY) 27,28 que é muito útil para discernir sinais devidos a solutos com coeficientes de difusão diferentes em uma mistura.
Apesar de todos estes benefícios, é necessário ter em mente que a amostra a ser analisada deve possuir uma concentração tal que seja possível obter um bom espectro de 1H em apenas um pulso (ex. > 0.01 g/mol.l utilizando um equipamento de 7.05 Tesla - 300 MHz para 1H). Caso contrário, as técnicas de RMN 2D usando gradiente de campo não oferecem nenhuma vantagem sobre aquelas que empregam o phase cycling, pois as dificuldades serão grandes para qualquer técnica 2D ou mesmo 1D. A concentração da amostra continua, apesar das evoluções técnicas, sendo um fator limitante para a determinação estrutural por RMN. Vale lembrar que para uma análise de RMN necessita-se de uma concentração, no mínimo, 10.000 vezes maior do que para uma análise de ultra-violeta (UV).
Em geral, a possibilidade da redução do tempo de análise de um experimento de RMN 2D por um fator de dez ou mais, assim como o restante das vantagens discutidas acima, significa que em um futuro próximo todos os equipamentos de RMN estarão providos dos acessórios de detecção inversa e gradiente de campo. Para muitos laboratórios, a aquisição destes acessórios para incorporação no equipamento já existente, pode ser feito sem maiores mudanças no hardware. Já para aqueles equipamentos mais antigos ( > 8 anos), esta mudança pode significar a completa troca do console, ou seja de quase todo o hardware, tornando-se pouco exeqüível devido ao elevado custo.
DETALHES TÉCNICOS
O johnstonol foi isolado e purificado no Laboratório de Produtos Naturais do Instituto de Química da UFRJ e apresenta-se como um sólido cristalino incolor. As medidas de RMN foram todas feitas em tubo de 5 mm de diâmetro com uma amostra de 7.2 mg em 0.6 ml de CDCl3 (12 mg/ml), contendo 0.1% v/v de TMS (referência), a 303 K e sem rotação (fundamental para 2D inverso). O equipamento de RMN utilizado nas análises foi um BRUKER DRX300 (300.13 MHz/1H e 75.47 MHz/13C, a 7.0463 Tesla), equipado com sonda inversa multinuclear (1H, 2H, 31P-107Ag) contendo três bobinas de gradiente (X,Y,Z; mas todos os experimentos aqui descritos podem ser executados apenas com a bobina do eixo Z) e uma unidade geradora de pulsos de gradiente de campo de amplitude máxima de 50 gauss/cm para cada eixo. Nos experimentos 1D (1H) e 2D (1H,1H e 1H,13C) foram utilizadas: para as aquisições, larguras espectrais SWH = 7.7 ppm (determinado no espectro de 1H) e SWC = 160 ppm (estimado), perídodo de relaxação de 1.3s (D1), pulso de 90o para 1H de 8.9ms com 6.0dB de potência (é conveniente calibrar à cada amostra) e para 13C de 14.0ms com 2.0dB de potência, pulsos senoidais de gradiente de 1ms de duração e 100 ms de recuperação; e para os processamentos matrizes de dados de 2048 x 1024 (F2 x F1) com zero filling e linear prediction1,5,29,30. Todos os programas de pulsos e de processamentos foram executados com o software XwinNmr 1.3/9.
gs-DQF-COSY: Foi utilizada na aquisição uma matriz de dados de 2048 x 256 com NS = 2 (number of scans ou número de varreduras em cada um dos 256 experimentos) e três pulsos de gradiente (PG) de intensidades 10:10:30 gauss/cm por eixo1,5,29,30.
HSQC-ED: Foi utilizada na aquisição uma matriz de dados de 2048 x 256 com NS = 2 e PG= 40:10:-40:10 gauss/cm em cada eixo, no modo fase sensitivo Echo/Antiecho-TPPI e desacoplado de carbono (GARP)1,5,16-20,29,30.
HSQC-TOCSY: Foi utilizada na aquisição uma matriz de dados de 2048 x 256 com NS = 4 e PG = 40:10:-40:10 gauss/cm em cada eixo, no modo fase sensitivo Echo/Antiecho-TPPI, desacoplado de carbono (GARP) e pulso gaussiano de excitação de 50ms com spin-lock de 2.5ms e 40 loops para o ciclo MLEV-161,5,15,16,21,29,30.
gs-HMBC: Foi utilizada uma matriz de dados de 2048 x 128 com NS=8 e PG=25:15:20 na aquisição. Note na Fig. 4 que devido ao "não desacoplamento de carbono" durante a aquisição e ao "baixo corte dos sinais 3D" para a obtenção dos contornos 2D, alguns sinais estão desdobrados de poucos (< 20) a vários Hz (> 150). O primeiro deve-se a diferença entre a constante de acoplamento média utilizada (8 Hz) para o mixing time (a a 1/J) e a real, que é maior, resultando em um dubleto de magnitude nJC,H. O segundo deve-se a imperfeições do filtro quântico (low pass filter) utilizado para eliminar os acoplamentos 1JC,H, e que não deve ser confundido com sinais devidos a nJC,H1,5,15,24,29,30 .
NOESY: Foi utilizada uma matriz de dados de 2048 x 256 com NS=32, com tempo de mistura (mixing time) de 1.0s, no modo fase sensitivo usando TPPI. É necessário otimizar o tempo de evolução dos spins (tempo de mistura), que é dependente do tempo de relaxação T1 (pode ser estimado por um rápido experimento de inversão-recuperação)1,2,3,5,29,30 .
AGRADECIMENTOS
Ao CNPq, CAPES, FINEP-PADCT I, FUJB e FAPERJ pelos auxílios financeiros recebidos. Em especial a Dra. Liane F. Pitombo pela amostra de johnstonol e ao Prof. Angelo da Cunha Pinto pela revisão e comentários sobre o texto.
- 1. Croasmun, W. R.; Carlson, R. M. K.; Eds; Two-Dimensional NMR Spectroscopy; VCH, New York, 1994, e referęncias citadas no texto.
- 2. Sanders, J. K. M.; Constable, E. C.; Hunter, B. K.; Modern NMR Spectroscopy, Oxford, New York, 1989.
- 3. Derome, A. E.; Modern NMR Techniques for Chemistry Research; Pergamon; Oxford, 1987.
- 4. Oschkinat, H.; Müller, T.; Dieckmann, T.; Angew. Chem., Int. Ed. Engl. 1994, 33, 277.
- 5. Braum, S.; Kalinowski, H. -O.; Berger, S.; 100 and More Basic NMR Experiments; VCH; Weinheim, 1996, e referęncias citadas no texto.
- 6. Breitmaier, E.; Voelter, W.; Carbon-13 NMR Spectroscopy; Verlag; Weinheim, 1987.
- 7. Silverstein, R. M.; Bassler, G. C.; Morrill, T. C.; Spectrometric Identification of Organic Compounds; Wiley & Sons; New York, 1995.
- 8. Sanders, J. K. M.; Mersh, J. D.; Prog. Nucl. Magn. Reson. Spectrosc. 1983, 15, 353.
- 9. Barfield, M.; Chakrabarti, B.; Chem. Rev. 1969, 69, 757.
- 10. Kaiser, C. R.; Rittner, R.; Basso, E. A.; Magn. Reson. Chem. 1997, 35, 609, e referęncias citadas no texto.
- 11. Kienlin, M.; Moomen, C. T. W.; Toorn, A.; Zijl, P. C. M.; J. Magn. Reson. 1991, 93, 423.
- 12. Bax. A., Freeman, R.; J. Magn. Reson. 1981, 44, 542.
- 13. Davis, A. L.; Laue, E. D.; Keeler, J; Moskau, D.; Lohman, J.; J. Magn. Reson. 1991, 94, 637.
- 14. Hurd, R. E.; J. Magn. Reson. 1990, 87, 422.
- 15. Wilker, W.; Leibfritz, D.; Kerssebaum, R.; Bermel, W.; Magn. Reson. Chem. 1993, 31, 287.
- 16. Kaiser, C. R.; Pitombo, L. F.; Pinto, A. C.; Spectroscopy Letters 1998, 31, 573.
- 17. Kay, L. E.; Bax, A.; J. Magn. Reson. 1989, 84, 598.
- 18. Schmidt, J. M.; Rüterjans, H.; J. Am. Chem. Soc. 1990, 112, 1279.
- 19. Kontaxis, G.; Stonehouse, J.; Laue, E. D. ; Keeler, J.; J. Magn. Res. Ser. A 1994, 111, 70.
- 20. León, I.; Enríquez, R. G.; McLean, S.; Reynolds, W. F.; Yu, M.; Magn. Reson. Chem. 1998, 36, S111.
- 21. Martin, G. E.; Spitzer, T. D.; Crouch, R. C.; Luo, J. -K.; Castle, R. N.; J. Heterocyclic Chem. 1992, 29, 577.
- 22. Pitombo, L. F.; Kaiser, C. R.; Pinto, A. C.; Bol. Soc. Chil. Quim. 1996, 41, 433.
- 23. Suzuki, M.; Segawa, M.; Suzuki, T.; Kurosawa, E.; Bull. Chem. Soc. Jpn. 1983, 56, 3824.
- 24. Bax, A.; Summers, M. F.; J. Am. Chem. Soc. 1986, 108, 2093.
- 25. Piotto, M.; Saudek, V.; Sklenar, V.; J. Biomol. NMR 1992, 2, 661.
- 26. Sklenar, V.; Piotto, M.; Leppik, R.; Saudek, V.; J. Magn. Reson., Series A 1993, 102, 241.
- 27. Wu, D.; Chen, A.; Johnson Jr.; C. S.; J. Magn. Reson., Series A 1995, 115, 260.
- 28. Altieri, A. S.; Hinton, D. P.; Byrd, R. A.; J. Am. Chem. Soc. 1995, 117, 7566.
- 29. Biegler, P.; NMR Spectroscopy: Processing Strategies, VCH, Weinheim, 1997.
- 30. Harris, R. K.; Kowalewski, J.; Menezes, S. C.; Pure & Appl. Chem. 1997, 69, 2489.
Datas de Publicação
-
Publicação nesta coleção
14 Out 2000 -
Data do Fascículo
Abr 2000
Histórico
-
Aceito
05 Jul 1999 -
Recebido
13 Jul 1998