Resumo
Hydrotalcite-like compounds having Mg partially replaced by Cu or Mn were prepared and used as precursors for two mixed oxides (Cu-OM50 and Mn-OM50) that were evaluated for SOx removal in the presence of O2, NO and CO. Under SO2/O2 reaction system, SOx removal was slightly higher over Cu-OM50. The addition of CO and NO to the feed markedly hindered the SO2 oxidation over Cu-OM50 while no significant effect was observed for Mn-OM50. For the regeneration step, the use of propane instead of H2 reduces regeneration capacity, mainly for Cu-OM50. Mn-OM50 was less affected by the feed composition, suggesting that it was a promising additive for SOx removal.
SOx emission control; sulfur-transfer catalysts; Mg,Al-mixed oxides
SOx emission control; sulfur-transfer catalysts; Mg,Al-mixed oxides
ARTIGO
Efeito da composição das correntes do conversor das unidades de FCC no desempenho catalítico de aditivos DESOx
Influence of the composition of the streams to the FCC converter unit on the catalytic performance of DESOx additives
Carla Maria Salerno PolatoI,# # Endereço atual: Instituto Nacional da Propriedade Industrial, Pça Mauá, 7, 22081-240 Rio de Janeiro - RJ, Brasil ; José Luiz Fontes MonteiroI; Cristiane Assumpção HenriquesII,* * e-mail: cah@uerj.br
IPrograma de Engenharia Química, COPPE, Universidade Federal do Rio de Janeiro, CP 68502, 21941-972 Rio de Janeiro - RJ, Brasil
IIInstituto de Química, Universidade do Estado do Rio de Janeiro, Rua São Francisco Xavier, 524, 20550-900 Rio de Janeiro - RJ, Brasil
ABSTRACT
Hydrotalcite-like compounds having Mg partially replaced by Cu or Mn were prepared and used as precursors for two mixed oxides (Cu-OM50 and Mn-OM50) that were evaluated for SOx removal in the presence of O2, NO and CO. Under SO2/O2 reaction system, SOx removal was slightly higher over Cu-OM50. The addition of CO and NO to the feed markedly hindered the SO2 oxidation over Cu-OM50 while no significant effect was observed for Mn-OM50. For the regeneration step, the use of propane instead of H2 reduces regeneration capacity, mainly for Cu-OM50. Mn-OM50 was less affected by the feed composition, suggesting that it was a promising additive for SOx removal.
Keywords: SOx emission control; sulfur-transfer catalysts; Mg,Al-mixed oxides/spinels.
INTRODUÇÃO
As recentes preocupações com as questões ambientais, que se traduzem em legislações governamentais mais rigorosas, bem como a tendência mundial de processamento de frações de petróleo cada vez mais pesadas e ricas em compostos sulfurados e nitrogenados, têm direcionado as pesquisas para a redução nas emissões de monóxido de carbono, óxidos de enxofre e óxidos de nitrogênio resultantes da queima do coque no regenerador das unidades de FCC (craqueamento catalítico fluido) existentes nas refinarias.
Uma alternativa para a redução das emissões de SOx é o uso de substâncias cataliticamente ativas em reações de oxidação como aditivos para os catalisadores de FCC. O uso de aditivos requer pouco investimento de capital, exceto pelo custo da carga de aditivo no sistema e pela disponibilidade de uma planta Claus para a recuperação do H2S gerado no processo. A boa performance de um catalisador para remoção de SOx encontra-se associada à sua atividade para promover os três tipos de reação que ocorrem no processo: oxidação do SO2 a SO3 nas condições de operação do regenerador (953-1003 K); quimissorção do SO3 na forma de sulfatos e, regeneração do catalisador por redução dos sulfatos sob a forma de H2S no riser (reator de leito móvel ascendente) (793-803 K).1
No que se refere à formulação do catalisador, os óxidos básicos são considerados os aditivos mais ativos para a fixação do SO3 sob a forma de sulfato. Entretanto, os sulfatos formados com os óxidos CaO e MgO são extremamente estáveis e dificilmente se decompõem na zona de reação. A alumina também pode ser usada, mas, neste caso, devido à instabilidade térmica do sulfato de alumínio nas condições de operação do regenerador das unidades de FCC, a quantidade de SO3 fixado na alumina é relativamente baixa. Assim sendo, compostos com basicidade intermediária, tais como óxidos mistos e espinélios de magnésio e alumínio, têm sido empregados de forma a encontrar um ponto ótimo entre a fixação de SO3 como sulfato e a regeneração do catalisador.2-5 Tanto os espinélios de magnésio e alumínio, estequiométricos (MgAl2O4) ou não estequiométricos (MgAl2O4.nMgO), como os óxidos mistos dos dois metais são obtidos a partir da calcinação de precursores à base de hidroxicarbonatos de magnésio e alumínio, pertencentes a uma classe de argilas aniônicas denominadas hidrotalcitas. Como o teor de SO3 no regenerador é relativamente baixo, faz-se necessária a adição de um outro componente que possua propriedades redox. Este componente promoveria a oxidação do SO2 a SO3 no regenerador bem como a recuperação, no riser, do óxido básico pela redução das espécies sulfatadas. As referências encontradas na literatura registram a incorporação de diferentes metais (Ce, Cu, Co e Fe, por exemplo) em compostos tipo hidrotalcita (HTLC) como a maneira de gerar as propriedades oxidantes e redutoras necessárias ao seu bom desempenho na remoção do SOx.2-4,6-12 Dentre estes metais, os catalisadores que apresentam cobre como promotor na sua composição têm se destacado dentre os vários sistemas estudados.2-4,10-12 A introdução de Mn aparece, também, como promissora, uma vez que catalisadores contendo óxidos de manganês são utilizados com eficiência em muitos processos industriais que envolvem reações de oxi-redução, devido aos diversos estados de oxidação (+2, +3, +4 e +7) que o elemento pode assumir. Particularmente com relação aos processos de remoção de SOx, o uso de catalisadores baseados em óxidos de manganês tem sido investigado em trapas para captura de SOx da corrente de gases de exaustão de motores diesel ou a gasolina, com o objetivo de aumentar a vida útil dos catalisadores automotivos.13,14 Por outro lado, no que concerne aos aditivos para catalisadores de FCC, o presente trabalho tem aspecto inovador, uma vez que não se encontra na literatura uma investigação sistemática do uso de aditivos contendo manganês.
No estudo realizado, óxidos mistos de Mg e Al com razão molar M3+/(M2+ + M3+) igual a 0,50, obtidos a partir de compostos tipo hidrotalcita contendo cobre ou manganês incorporados na sua estrutura, foram comparativamente avaliados como catalisadores para remoção de SOx. Na etapa de adsorção oxidativa do SO2, os testes foram realizados na presença de O2, NO e CO, sob condições que visam simular aquelas existentes na fase densa dos regeneradores das unidades de FCC. Para a etapa de regeneração do catalisador, que ocorre no riser, foram empregadas comparativamente duas correntes distintas, uma contendo 30% de H2/He e outra contendo 30% de C3H8/He.
PARTE EXPERIMENTAL
Preparação dos catalisadores
Os compostos tipo hidrotalcita (HTLCs) precursores dos óxidos mistos estudados como catalisadores foram sintetizados por coprecipitação a temperatura ambiente. Duas soluções, A e B, foram utilizadas na preparação do gel de síntese, cuja composição foi estabelecida a partir da fórmula geral 0,25 M(NO3)2:1,25 Mg(NO3)2:1,5 Al(NO3)3:2 Na2CO3: 7,5NaOH, sendo M = Cu ou Mn. Uma amostra de hidrotalcita contendo apenas Mg e Al foi também preparada para servir como referência na caracterização físico-química das demais amostras sintetizadas. A solução A era preparada a partir da dissolução dos nitratos dos metais em 100 mL de água deionizada de modo a se obter a composição desejada para o gel de síntese ([Al] + [Mg] + [M] = 1,5 mol/L; [Al]/([Al] + [Mg] + [M]) = 0,50; [Mg]/[M] = 5). No preparo da solução B, Na2CO3 e NaOH eram dissolvidos em água deionizada (100 mL) de modo a se obter uma concentração de carbonato igual a 1 mol/L e de NaOH igual a 3,75 mol/L.
De acordo com o procedimento de síntese, a solução A era gradativamente adicionada à solução B, sob agitação. O gel era, então, envelhecido por 18 h, em estufa, a 333 K e, posteriormente, filtrado e lavado com água destilada a quente (temperatura entre 353 e 363 K) até que a água de lavagem apresentasse pH = 7. O material obtido era seco em estufa, a 353 K, por 12 h. As amostras assim obtidas foram denominadas pela sigla M-HT50, em que M indica o metal que substitui parcialmente o Mg na estrutura do composto tipo hidrotalcita (Cu ou Mn).
Os óxidos mistos e/ou espinélios de Mg e Al, contendo ainda Cu ou Mn, foram obtidos por decomposição térmica dos HTLCs precursores sob fluxo de ar (100 mL/min) da temperatura ambiente até 1023 K, a uma taxa de aquecimento de 10 K/min, sendo, a seguir, mantidas nesta temperatura por 2 h. Estas amostras foram denominadas pela sigla M-OM50 (OM = óxido misto), em que M representa Cu ou Mn.
Caracterização físico-química dos catalisadores
As amostras foram caracterizadas por difração de raios X, para a identificação da(s) fase(s) presente(s) nos precursores e nos óxidos mistos/espinélios obtidos pelo tratamento térmico dos HTLCs, em um difratômetro Miniflex-Rigaku, empregando-se radiação Cu Kα em uma faixa de 2 de 10 a 80º. A composição química foi determinada por fluorescência de raios X, em espectrômetro Rigaku, modelo Rix 3100, controlado por computador através do software Rix 3100 e dotado de tubo gerador de raios X de Rh. A contagem dos pulsos foi feita através de um detector proporcional de fluxo.
As amostras de hidrotalcitas sintetizadas foram submetidas às análises termodiferencial (ATD) e termogravimétrica (ATG), a fim de determinar as perdas de massa associadas às etapas de desidratação e desidroxilação/descarbonatação. A análise foi realizada em termobalança Rigaku modelo PAS100, sob fluxo de ar seco (30 mL/min), numa taxa de aquecimento de 10 K/min até 1173 K.
A análise textural das amostras calcinadas foi realizada no equipamento ASAP (Accelerated Surface Area and Porosity) modelo 2010 da Micromeritics. Este equipamento fornece, a partir das medidas de adsorção e dessorção do N2 a 77 K, a área específica BET, a área e o volume de microporos pelo método t (usando a Equação de Harkins e Jura) e a área, o volume e a distribuição de mesoporos pelo método BJH.
Avaliação catalítica
A reação foi conduzida em um micro-reator de quartzo sob pressão atmosférica. Para a etapa de adsorção oxidativa, a temperatura de reação foi fixada em 993 K e o tempo em 10 min, tendo sido empregadas três correntes de composição distintas: 1630 ppm de SO2, 1,6% (v/v) de O2 e balanço de He; 1630 ppm de SO2, 1,6% (v/v) de O2, 5% (v/v) de CO e balanço de He; 1630 ppm de SO2, 1,6% (v/v) de O2, 5% (v/v) de CO, 2630 ppm de NO e balanço de He. Para a etapa de redução do sulfato formado, foram utilizadas duas correntes com diferentes composições: 30% de H2 em He ou 30% de C3H8 em He. A temperatura de redução foi fixada em 803 K, por 30 min, e, após este tempo, a temperatura de reação era elevada até 1073 K, numa taxa de aquecimento de 10 K/min.
Os produtos de reação foram analisados em linha através de um espectrômetro de massas quadrupolo da Balzers, modelo Prisma-QMS 200, monitorando-se H2 (m/z = 2), H2O (m/z = 18), O2 (m/z = 16, 32), H2S (m/z = 32, 33, 34), SO2 (m/z = 32, 48, 64), CO (m/z = 28), CO2 (m/z = 44), NO (m/z = 30) e NO2 (m/z = 30, 46). As análises quantitativas foram baseadas nas áreas dos picos dos diversos componentes e seus perfis de fragmentação, com o uso de fatores de calibração e de deconvolução numérica.
RESULTADOS E DISCUSSÃO
Caracterização físico-química dos catalisadores
A Tabela 1 apresenta os resultados de caracterização química (FRX) dos compostos tipo hidrotalcita sintetizados. Os resultados mostraram-se compatíveis com a composição dos géis de síntese correspondentes.
Os difratogramas de raios X dos compostos tipo hidrotalcita são apresentados na Figura 1. Para as três amostras observou-se a presença dos picos característicos da fase hidrotalcita na forma carbonato com estrutura lamelar e alguns picos de difração correspondentes a uma outra fase, identificada como hidróxido de alumínio, Al(OH)3-bayerita.15 Nos compostos tipo hidrotalcita sintetizados com razão Al/(Al + Mg) acima de 0,33 ocorre um aumento no número de íons Al3+ vizinhos na camada de hidróxidos. Este efeito se sobrepõe à repulsão provocada pelas cargas positivas que determinaria seu afastamento e leva à segregação da fase Al(OH)3.16 Não foi observada segregação das fases de óxidos de cobre ou de manganês, o que sugere que estes elementos tenham sido incorporados na estrutura lamelar do HTLC. Esta proposta é reforçada pela comparação dos parâmetros de rede da fase lamelar presente nas amostras Cu-HT50 e Mn-HT50, que foram diferentes daqueles da fase lamelar contendo apenas Mg e Al (amostra HT50).
Os perfis de ATG e ATD correspondentes às amostras nas quais o Mg foi parcialmente substituído por Cu ou Mn apresentaram as duas perdas de massa típicas dos HTLCs na forma carbonato, ambas associadas a transformações endotérmicas. A primeira (400-500 K), relacionada à perda de água interlamelar, e a segunda (550-700 K), à desidratação e descarbonatação, com liberação de H2O e CO2.16 A perda de massa observada em temperaturas intermediárias (na faixa dos 523 K) pode ser associada à desidroxilação da fase bayerita presente em todas as amostras. Os resultados das análises termogravimétricas das amostras sintetizadas são apresentados na Tabela 2.
A incorporação do metal de transição influenciou os resultados de ATG/ATD, quando comparados aos do HTLC contendo apenas Mg e Al (HT50), tendo sido observada uma leve redução nas perdas de massa associadas às duas transformações de fase, bem como um decréscimo na temperatura na qual a taxa máxima de cada transformação foi observada, efeito este mais marcante para a mostra contendo Mn.
Como mostrado pelos difratogramas de raios X apresentados na Figura 2, para a amostra HT50, o tratamento térmico a 1023 K determinou a segregação de uma fase óxido misto (Mg(Al)O) de baixa cristalinidade com estrutura do tipo MgO-periclásio,15 além da formação de γ-Al2O315 proveniente da decomposição da bayerita. Para a amostra Cu-HT50, a calcinação sob fluxo de ar a 1023 K ocasionou a destruição da estrutura lamelar e levou à formação, como no caso da amostra HT50, de um óxido misto de baixa cristalinidade (Mg(Cu,Al)O) com estrutura tipo MgO-periclásio.15 Além desta fase, observou-se também a formação de uma outra fase pouco cristalina, associada ao espinélio MgAl2O4.15 No caso da amostra contendo Mn (amostra Mn-HT50), o tratamento térmico levou à formação de espinélios de Mn, Mg e Al com estrutura cúbica15 e baixa cristalinidade.
A Tabela 3 apresenta os principais resultados de caracterização textural dos óxidos mistos obtidos a partir dos compostos tipo hidrotalcita. A análise das isotermas de adsorção/dessorção de N2 a 77 K das amostras calcinadas permitiu classificá-las como do tipo IV (segundo IUPAC), indicando serem as mesmas correspondentes a sólidos mesoporosos.
Avaliação dos aditivos para remoção de SOx
1ª etapa: adsorção oxidativa do SO2
Na etapa de adsorção oxidativa de SO2 as amostras foram submetidas a três correntes de composições distintas, visando avaliar a influência da presença de NO e CO na eficiência de remoção de SOx, durante a etapa de queima do coque que ocorre no regenerador das unidades de FCC.
Para uma corrente de sulfatação composta apenas por SO2 e O2 (corrente 1), os resultados da etapa de adsorção oxidativa do SOx, mostrados nas Tabelas 4 e 5, indicam que, em relação ao óxido misto de Mg e Al (amostra OM50), a quantidade de SOx capturada pelo catalisador foi incrementada pela incorporação de Mn e, principalmente, de Cu na estrutura do HTLC precursor, sendo este comportamento catalítico relacionado às propriedades redox destes metais de transição, que atuam, assim, na promoção da oxidação do SO2 a SO3. A amostra OM50 foi capaz de capturar apenas 381 µmol/g de SO2, enquanto as amostras Cu-OM50 e Mn-OM50 removeram, respectivamente, 3530 e 2890 µmol/g.
Os cálculos referentes à eficiência de remoção de SOx (definida como a relação entre o número de mols de SO2 removido e o número de mols de óxido de magnésio presentes no catalisador) indicaram que aproximadamente 34% dos sítios de quimissorção presentes na amostra Mn-OM50 foram utilizados, enquanto na Cu-OM50 este número correspondeu a 38% dos sítios. Estes valores de eficiência de remoção de SOx foram calculados admitindo-se que apenas o magnésio presente no óxido misto participa da formação de sulfato (MgSO4), durante a etapa de adsorção oxidativa do SOx. O alumínio não foi incluído neste cálculo em função da instabilidade térmica do Al2(SO4)3 nas condições de reação. Cu e Mn também não foram incluídos neste cálculo devido ao fato das fases MnSO4 e CuSO4 não terem sido identificadas nos estudos de difração de raios X das amostras sulfatadas.17
A análise comparativa dos dados referentes ao consumo de SO2 quando os catalisadores foram submetidos à corrente 1 (SO2 + O2) indicou que o Cu promoveu a captura de SOx de modo mais eficiente que o Mn. Entretanto, quando a composição da corrente do regenerador se tornou mais próxima da condição real, observou-se que a presença de CO e, principalmente, de NO na corrente de sulfatação alterou de modo significativo a eficiência de remoção de SOx da amostra Cu-OM50, que decresceu abruptamente para a corrente de sulfatação composta de SO2, CO, NO, e O2, conforme mostrado na Tabela 4. Ao simular as condições da fase densa do regenerador das unidades de FCC, onde a concentração de CO é muito maior que a concentração de O2,18 observou-se que, introduzindo-se apenas CO no sistema SO2 + O2, ocorreu uma redução no consumo de SO2 e uma elevada conversão de CO. Dada a quantidade insuficiente de oxigênio para a oxidação completa de ambos os reagentes, os resultados encontrados permitem sugerir que a oxidação do CO tenha ocorrido preferencialmente sobre o catalisador estudado. Isto porque o valor obtido experimentalmente para a conversão de CO (Tabela 4) foi superior ao previsto pela estequiometria da reação considerando-se as quantidades de CO e O2 presentes no meio reacional, sugerindo, assim, que parte do consumo de CO e do SO2 poderia estar associado à reação:

como evidenciado pela presença de enxofre condensado na saída do reator, ou ainda à redução das espécies de Cu(II) existentes no catalisador.17
No caso da presença de NO, este competiria com o SO2 pelos mesmos sítios ativos.18-20 Desta forma, poder-se-ia especular que, em presença de NO, a reação de oxidação do SO2 tenha ocorrido em menor extensão pelo fato dos sítios ativos catalisarem preferencialmente a redução do NO, que experimentou uma conversão de 70%.

Assim, óxidos mistos derivados de compostos tipo HTLCs contendo Cu em sua composição parecem ser aditivos eficientes para catalisar a reação entre NOx e CO ou coque (redução do NOx pelo CO ou pelo coque formando N2 e CO2) na fase densa de regenerador, sem, contudo, serem muito ativos para a remoção simultâneade SOx. Este resultado contraria a proposta feita por Corma et al.4 ao avaliarem óxidos mistos derivados de Cu-HTLCs. Segundo estes autores, as amostras contendo cobre seriam promissoras para a remoção combinada de SOx e NOx. Entretanto, esta proposição foi feita sem que os autores4 tivessem estudado a remoção simultânea destes dois poluentes atmosféricos. Como visto no presente trabalho, a amostra Cu-OM50 pode ser considerada o melhor aditivo para a remoção de SOx, quando avaliada na presença de uma corrente composta apenas por SO2 e O2. No caso de uma corrente com composição mais próxima daquela encontrada nas UFCC (unidades de craqueamento catalítico em leito fluido), as considerações feitas por Corma e colaboradores4 falham, pois a capacidade de remoção de SOx da amostra contendo cobre mostrou-se extremamente influenciada pela presença de CO e principalmente de NOx no meio reacional.
Wen et al.19,20 avaliaram óxidos mistos derivados de HTLCs nos quais o Mg foi substituído por Cu na remoção de NOx em presença de SO2. Observaram um decréscimo significativo da conversão de NO devido à presença do SO2 na carga, que causaria um rápido envenenamento do aditivo. Embora os autores tenham empregado condições que simulassem a fase densa dos regeneradores das unidades de FCC (corrente composta por SO2, NO, CO e O2), as condições de reação e as técnicas empregadas para a avaliação do desempenho catalítico dos aditivos foram diferentes daquelas usadas no presente trabalho, de modo que os resultados obtidos não puderam ser diretamente comparados entre si. Wen et al.19,20 realizaram, também, estudos por espectroscopia na região do infra-vermelho dos aditivos contendo cobre. Os resultados indicaram que as espécies Cu(I) seriam os sítios ativos para a quimissorção do CO, enquanto que as espécies Cu(II) seriam ativas para a adsorção do NO, mas não do CO. Segundo os autores, a competição entre SO2 e NO pelos sítios Cu(II) seria a causa da desativação observada.
Estudos termodinâmicos envolvendo as múltiplas reações associadas ao processo de remoção catalítica do SOx foram realizados por Pereira et al..21 Seus resultados indicaram que a redução do CuO a Cu2O seria favorecida termodinamicamente mesmo em condições oxidantes. Neste caso, o CuO desempenharia papel ativo na etapa de oxidação do SO2, como sugerido a seguir:

Desta forma, no caso da corrente composta por SO2, NO, CO e O2, existiria uma competição entre o SO2 e o NO pelas espécies Cu(II), que seriam sítios ativos tanto para a oxidação do SO2 quanto para a quimissorção do NO a ser reduzido pelo CO. Nas condições estudadas neste trabalho, os resultados sugerem que a reação de redução do NO ocorre preferencialmente à de oxidação do SO2.
Para a amostra Mn-OM50, as alterações na composição da corrente de sulfatação influenciaram de modo pouco significativo a remoção de SO2, como mostrado na Tabela 5. Constata-se, assim, que este catalisador se caracteriza por ser ativo e altamente seletivo para aoxidação do SO2, sendo, porém, muito pouco ativo para catalisar reações envolvendo CO e NO.
No caso do aditivo contendo manganês, o estudo termodinâmico realizado por Pereira et al.21 previu que, a 993 K e em presença de atmosfera oxidante, o MnO seria oxidado a Mn2O3, que, por sua vez, reagiria em grande extensão com o SO2 formando o MnSO4:

Muito embora a formação de MnSO4 não tenha sido observada por difração de raios X no aditivo Mn-OM50 após a etapa de adsorção oxidativa, a presença da fase MnS foi identificada no difratograma de raios X do aditivo regenerado.17 Este fato pode ser considerado uma forte evidência da formação do MnSO4, possivelmente sob a forma de partículas muito pequenas, não detectáveis por DRX, tendo em vista ele formar MnS por redução em presença de H2.
A coexistência das espécies Mn(III) e Mn(II) na amostra Mn-OM50 foi também sugerida por análises por espectroscopia de reflectância difusa no UV-VIS.17 A comparação dos espectros da amostra antes e após a etapa de adsorção oxidativa mostrou um decréscimo na intensidade da banda associada ao Mn(III) com a sulfatação da amostra, o que estaria relacionado à formação do MnSO4. A formação de MnS (indicada pela análise por difração de raios X), por redução do MnSO4, explicaria a menor intensidade da banda correspondente ao Mn(III) no espectro de reflectância difusa da amostra regenerada quando comprado ao espectro da amostra calcinada.
Deste modo, tendo o manganês um papel ativo na captura do sulfato (não apenas como promotor de oxidação, mas também como sítio de fixação do SO3), restariam poucos sítios disponíveis para promover a reação entre o CO e o NO.
2ª etapa: regeneração do catalisador
A regeneração dos catalisadores sulfatados a partir da corrente de sulfatação 1 (SO2 + O2) foi estudada, buscando-se avaliar, também, o efeito da composição da corrente de regeneração (30% H2/He ou 30% C3H8/He).
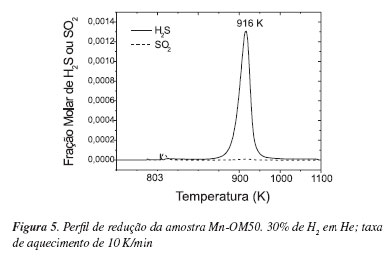
Independentemente da composição da corrente de regeneração, quando foi empregada temperatura similar àquela de operação do riser das unidades de FCC (803 K) não foi observada a regeneração dos catalisadores sulfatados. Em função disto, foram realizados testes de TPR/MS, com aquecimento contínuo até 1073 K, cujos resultados são apresentados nas Figuras 3 e 4, para a amostra Cu-OM50, e 5 e 6, para a amostra Mn-OM50. As análises quantitativas são apresentadas na Tabela 6.
Os resultados obtidos para a amostra Cu-OM50 indicaram a completa regeneração da amostra em presença da corrente de H2/He. O H2S foi o principal produto da redução do sulfato, sendo pequena a fração reduzida parcialmente (associada à formação de SO2). A avaliação do perfil de redução da amostra (Figura 3) indica que a temperatura inicial para a liberação de H2S foi superior a 850 K e, portanto, maior que a temperatura de reação praticada no riser da unidade de FCC. A liberação do SO2 foi simultânea à do H2S.
A substituição do H2 pelo C3H8 reduziu de modo significativo a regeneração do catalisador. Além disso, a análise da Figura 4 mostra um leve deslocamento do perfil de liberação do H2S para temperatura mais elevada (Tmax desloca-se de 950 para 960 K) e um aumento da quantidade relativa de SO2 (redução parcial do sulfato), que passou a ser liberado, em sua maior parte, a temperaturas superiores a 1000 K. Estes resultados indicaram ser a redução do sulfato fortemente influenciada pela composição da corrente do riser.
A regeneração da amostra Mn-OM50, na presença de H2, levou à liberação simultânea de SO2 e de H2S, que, também neste caso, foi liberado a temperatura superior àquela encontrada no riser da unidade de FCC (Figura 5). No caso desta amostra, como não foi descartada a possibilidade de formação de MnSO4, este poderia ser reduzido a sulfeto nas condições da reação, contribuindo também para a liberação de SO2, de acordo com a reação apresentada a seguir:

As análises por difração de raios X da amostra confirmaram a formação da fase MnS (alabandita) no catalisador após a etapa de redução, sendo possível observar, também, que a estrutura tipo espinélio Mn-Mg-Al não foi recuperada, ocorrendo alterações estruturais que levaram à formação da fase MgO, com estrutura do tipo periclásio, e do espinélio MgAl2O4.17
A Figura 6 mostra que, em presença do C3H8, o perfil de liberação do H2S foi levemente deslocado para temperaturas maiores (Tmax desloca-se de 916 para 933 K). Observa-se também que o uso do C3H8 na corrente de regeneração aumentou a quantidade de enxofre liberado como SO2 e deslocou a faixa de temperatura na qual ocorre a liberação deste composto para valores muito maiores (> 1000 K). Tendências semelhantes foram observadas para a amostra contendo cobre.
A substituição do H2 pelo C3H8 reduziu a porcentagem de regeneração da amostra Mn-OM50 de modo menos significativo (de 58 para 41%) que o observado para a amostra Cu-OM50 (100 para 57%), o que indicaria ser o aditivo contendo manganês em sua composição menos susceptível a alterações na composição da corrente de redução no riser das unidades de FCC. Além disso, cabe ressaltar que tanto a temperatura inicial quanto a temperatura máxima de liberação de H2S foram menores que aquelas observadas para a amostra Cu-OM50, independentemente da atmosfera de redução estudada.
A avaliação global dos resultados indicou que o propano é um redutor menos eficiente que o hidrogênio, dados os menores níveis de redução observados para os dois catalisadores e também a maior liberação de SOx, indesejável nesta etapa do processo. Tendências similares foram observadas para o catalisador de CeO2 impregnado em óxido misto de Mg e Al (Al/(Mg+Al) = 0,50).18
Além disso, a diferença observada entre os catalisadores com relação aos níveis de redução alcançados concorda com a proposta de Kim e Juskelis6 de que a regeneração do catalisador seria determinada não apenas pela natureza da corrente de redução, como também pela composição do mesmo. Entretanto, os resultados obtidos contrariam o mecanismo proposto por aqueles autores,6 segundo o qual a redução do S6+ a S2- ocorreria por uma seqüência de reações consecutivas, tendo o SO2 como intermediário. Isto porque, em presença de C3H8 como agente redutor, foi possível observar claramente a liberação da maior parte do SO2 após a formação de H2S (Figuras 4 e 6). Além disso, quando o H2 foi usado como agente redutor, os perfis de redução das amostras Cu-OM50 e Mn-OM50 (Figuras 3 e 5, respectivamente) indicam que a liberação do SO2 ocorreu simultaneamente à do H2S.
CONCLUSÕES
Óxidos mistos/espinélios derivados de compostos tipo hidrotalcita (HTLC) com uma razão molar Al/(M2+ + Al) igual a 0,50 nos quais o Mg foi parcialmente substituído por Cu ou Mn foram preparados e avaliados como aditivos a serem incorporados aos catalisadores com o objetivo de reduzir as emissões de SOx nas unidades de FCC. Eles se mostraram mais eficientes para captura do SOx que o óxido misto contendo apenas Mg e Al, o que confirma que a presença de um componente com propriedades redox é essencial para a performance adequada de um aditivo DeSOx.
A comparação da quantidade de SOx capturado pelos aditivos Cu-OM50 e Mn-OM50, no caso da corrente composta apenas por SO2 + O2, indicou que a presença do cobre promoveu a captura de SOx de modo mais eficiente que a presença do manganês. Entretanto, a introdução de CO no sistema SO2 + O2 determinou uma redução na captura de SO2 e uma elevada conversão de CO, no caso da amostra Cu-OM50. Tendo em vista que o nível de conversão do CO foi superior ao previsto pela estequiometria da reação, considerando-se as quantidades de CO e O2 no meio reacional, acredita-se que parte do consumo do CO e do SO2 tenha sido devido à reação entre ambos formando CO2 e enxofre ou ainda à redução das espécies de Cu(II) existentes no catalisador. No caso da presença de NO e CO na corrente de reação, a captura do SO2 diminuiu significativamente, enquanto a reação entre CO e NO atingiu nível de conversão elevado. Existiria, assim, uma competição entre NO e SO2 pelos mesmos sítios ativos, espécies Cu(II), que catalisariam preferencialmente as transformações do NO. Deste modo, óxidos mistos derivados de compostos tipo HTLCs contendo Cu, Mg e Al em sua composição parecem ser aditivos eficientes para catalisar a reação entre NOx e CO na fase densa de regenerador, sem contudo serem muito ativos para remover simultaneamente o SOx.
A amostra Mn-OM50, por outro lado, mostrou-se um catalisador ativo e altamente seletivo para a remoção do SO2 da corrente de efluentes nas unidades de FCC, já que, diferentemente do observado para a amostra Cu-OM50, a capacidade de remoção de SO2 da amostra contendo manganês não foi afetada pela presença de CO e, principalmente, de NO na corrente de sulfatação. No catalisador contendo manganês, este desempenharia um papel ativo não apenas como promotor de oxidação, mas também na captura do sulfato (atuaria como sítio de fixação do SO3), de modo que restariam poucos sítios disponíveis para promover a reação entre CO e NO.
Com relação à etapa de regeneração dos catalisadores sulfatados, que foi avaliada na presença de diferentes agentes de redução (H2 e C3H8), embora o aditivo contendo manganês em sua composição tenha apresentado um nível de regeneração menor, ele mostrou-se menos susceptível a alterações na corrente de redução. Além disso, deve ser considerado, ainda, que na amostra Mn-OM50 tanto a temperatura inicial quanto a temperatura máxima de liberação de H2S foram menores que aquelas observadas para a amostra Cu-OM50, independentemente da atmosfera de redução estudada.
O uso de um hidrocarboneto (C3H8) como agente redutor diminuiu a regeneração dos aditivos estudados e aumentou a fração de sulfato reduzida parcialmente, aumentando, conseqüentemente, a indesejável liberação de SO2.
Conclui-se, assim, que a redução das espécies sulfatadas é influenciada tanto pela composição da corrente de redução quanto pelo metal de transição presente no catalisador.
MATERIAL SUPLEMENTAR
Está disponível em http://quimicanova.sbq.org.br, na forma de arquivo .PDF, com acesso livre. São apresentados os perfis de captura do SOx presente nas diferentes correntes de alimentação pelos aditivos avaliados.
AGRADECIMENTOS
À CAPES pela bolsa de Doutorado concedida a C. M. S. Polato e ao Programa PROCIÊNCIA da UERJ.
Recebido em 3/11/07; aceito em 22/7/08; publicado na web em 18/12/08
Material Suplementar

Figura 1S - clique para ampliar

Figura 2S - clique para ampliar
- 1. Cheng, W.-C.; Kim, G.; Peters, A. W.; Zhao, X.; Rajagopalan, K.; Catal. Rev. Sci. Eng 1998, 40, 39.
- 2. Palomares, A. E.; Lópes-Nieto, J. M.; Lázaro, F. J.; Lopes, A.; Corma, A.; Appl. Catal.,B 1999, 20, 257.
- 3. Corma, A.; Palomares, A. E.; Rey, F.; Appl. Catal.,B 1994, 4, 29.
- 4. Corma, A.; Palomares, A. E.; Rey, F.; Márques, F.; J. Catal 1997, 170, 140.
- 5. Wang, J. A.; Chen, L. F.; Ballesteros, R.; Montoya, A.; Domínguez, J. M.; J. Mol. Catal. A: Chem. 2003, 194, 181.
- 6. Kim, G.; Juskelis, M. V.; Stud. Surf. Sci. Catal 1996, 101, 137.
- 7. Trovarelli, A.; Leitenburg, C.; Borao, M.; Dolcetti, G.; Catal. Today 1999, 50, 353.
- 8. Polato, C. M. S.; Henriques, C. A.; Alcover Neto, A.; Monteiro, J. L. F.; J. Mol. Catal. A: Chem. 2005, 241, 184.
- 9. Wang, J. A.; Zhu, C. L.; Li, J.; J. Mol. Catal. A: Chem. 1999, 139, 31.
- 10. Centi, G.; Perathoner, S.; Catal. Today 2007, 127, 219.
- 11. Centi, G.; Perathoner, S.; Appl. Catal., B 2007, 70, 172.
- 12. Polato, C. M. S.; Henriques, C. A.; Rodrigues, A. C. C.; Monteiro, J. L. F.; Catal. Today 2008, 133-135, 534.
- 13. Li, L.; King, D. L.; Ind. Eng. Chem. Res 2005, 44, 168.
- 14. Tikhomirov, K.; Kröcher, O.; Elsener, M.; Widmer, M.; Wokaun, A.; Appl. Catal., B 2006, 67, 160.
-
15ICDD PDF-2 Database (Release 1998) - International Centre for Diffraction Data (ICDD). 12 Campus Boulevard Newton Square, Pennsylvania 19073-3273 USA.
- 16. Cavani, F.; Trifiró, F.; Vaccari, A.; Catal. Today 1991, 11, 173.
- 17. Polato, C. M. S.; Tese de Doutorado, Universidade Federal do Rio de Janeiro, Brasil, 2005.
- 18. Wen, B.; He, M.-Y.; Schrum, E.; Li, C.; J. Mol. Catal. A: Chem. 2002, 180, 187.
- 19. Wen, B.; He, M.-Y.; Appl. Catal., B 2002, 37, 75.
- 20. Wen, B.; He, M.-Y.; Costello, C.; Energy Fuels 2002, 16, 1048.
- 21. Pereira, H. B.; Polato, C. M. S.; Rodrigues, A. C. C.; Paredes, M. L. L.; Monteiro, J. L. F.; Henriques, C. A.; Anais do 14ş Congresso Brasileiro de Catálise, Porto de Galinhas, Brasil, 2007, CDRom.
Datas de Publicação
-
Publicação nesta coleção
26 Fev 2009 -
Data do Fascículo
2009
Histórico
-
Aceito
22 Jul 2008 -
Recebido
03 Nov 2007