Resumo
Advanced oxidative processes (AOPs) are based on chemical processes that can generate free radicals, such as hydroxyl radicals (.OH) which are strong, non-selective oxidant species that react with the vast majority of organic compounds. Nanostructured semiconductors, especially titanium dioxide (TiO2) in the anatase phase, are well-established photocatalysts for this process, which have proved to be useful in the degradation of dyes, pesticides and other contaminants. Research in different strategies for the synthesis of nanostructured semiconductors, with particular characteristic is currently a topic of interest in many studies. Thus, this paper presents a review about various synthesis strategies of nanostructured photocatalysts.
synthesis; nanostructured photocatalysts; photodegradation
synthesis; nanostructured photocatalysts; photodegradation
REVISÃO
Nanoestruturas em fotocatálise: uma revisão sobre estratégias de síntese de fotocatalisadores em escala nanométrica
Nanostructures in photocatalysis: a review about synthesis strategies of photocatalysts in nanometric size
Henrique A. J. L. MourãoI; Vagner R. de MendonçaI; Andréa R. MalaguttiII; Caue RibeiroII,* * e-mail: caue@cnpdia.embrapa.br
IDepartamento de Química, Universidade Federal de São Carlos, Rod. Washington Luiz, km 235, 13565-905 São Carlos - SP, Brasil
IIEMBRAPA Instrumentação Agropecuária, CP 741, 13560-970 São Carlos - SP, Brasil
ABSTRACT
Advanced oxidative processes (AOPs) are based on chemical processes that can generate free radicals, such as hydroxyl radicals (.OH) which are strong, non-selective oxidant species that react with the vast majority of organic compounds. Nanostructured semiconductors, especially titanium dioxide (TiO2) in the anatase phase, are well-established photocatalysts for this process, which have proved to be useful in the degradation of dyes, pesticides and other contaminants. Research in different strategies for the synthesis of nanostructured semiconductors, with particular characteristic is currently a topic of interest in many studies. Thus, this paper presents a review about various synthesis strategies of nanostructured photocatalysts.
Keywords: synthesis; nanostructured photocatalysts; photodegradation.
INTRODUÇÃO
Diversos setores da atividade industrial ou agrícola geram grandes quantidades de resíduos tóxicos ou ricos em atividade microbiana. Por exemplo, cerca de 30% dos corantes sintéticos (azocorantes) utilizados industrialmente são transformados em resíduos, uma vez que apresentam baixa fixação em fibras celulósicas.1 Outro exemplo de grande interesse encontra-se na contaminação de águas de irrigação agrícola por pesticidas e fertilizantes químicos (agroquímicos). Atualmente estas classes de compostos químicos são consideradas a segunda maior fonte de contaminação da água potável nos países em desenvolvimento.2 Por fim, efluentes com atividade microbiana também são ainda um grande problema quando se trata de saúde pública.34-5
Por diversas questões, cada vez mais as autoridades governamentais têm-se preocupado com os problemas ambientais gerados a partir destas atividades. O Brasil é pioneiro na edição de normas regulamentadoras para o descarte de efluentes líquidos, como exemplifica a Instrução Normativa Nº 2, de 3 de janeiro de 2008, do MAPA (Ministério da Agricultura, Pecuária e Abastecimento). Trata-se de novas normas de trabalho a serem seguidas pela aviação agrícola em conformidade com os padrões técnicos operacionais e de segurança para este setor.6 Estabeleceu-se que os eventuais restos de agrotóxicos remanescentes nos aviões utilizados na aplicação deverão passar para um sistema de oxidação de agrotóxicos, para redução da concentração de potenciais poluentes. Apesar de o gerador de ozônio (ozonizador) ser o sistema de oxidação citado nesta instrução normativa, novas estratégias de oxidação que apresentarem comprovado potencial podem ser utilizadas para este fim, como também poderão ser utilizadas em outros processos de descontaminação como, por exemplo, na despoluição de áreas contaminadas com corantes, atividade microbiana, entre outros.
Atualmente, os Processos Oxidativos Avançados (POAs) vêm sendo amplamente utilizados como uma alternativa no tratamento efluentes contaminados, em especial processos fotocatalíticos utilizados no tratamento de águas contaminadas por compostos orgânicos.7 Apesar de algumas peculiaridades, os mecanismos de ação dos POAs no processo de degradação dos contaminantes já são bem conhecidos. É notável, porém, que a investigação dos processos de síntese dos materiais utilizados nos POAs, assim como estudos sobre os efeitos inerentes às estratégias de síntese nos processos fotocatalíticos, ainda tenham sido pouco explorados.
Quando se utiliza POAs através de sistemas heterogêneos (como, por exemplo, semicondutores nanoparticulados em suspensão), a efetividade do processo fotocatalítico - visto corresponder a um processo de catálise heterogênea - dependerá essencialmente da área superficial do semicondutor, que é variável diretamente ligada ao tamanho da partícula utilizada.8 Desta forma, fotocatalisadores em escala nanométrica são bastante interessantes, pois tais materiais possuem alta área superficial específica.
Desta forma, esta revisão tem como objetivo principal discutir os principais métodos de síntese utilizados na obtenção de nanoestruturas com potencial fotocatalítico. Para isso, primeiramente serão discutidos os principais mecanismos de fotodegradação de moléculas orgânicas. Logo após, serão discutidos os principais métodos de síntese de fotocatalisadores em escala nanométrica. Posteriormente, serão apresentados vários exemplos de síntese de semicondutores na forma de nanopartículas e filmes finos imobilizados.
MECANISMO DE DEGRADAÇÃO DE MOLÉCULAS ORGÂNICAS
Nesta seção, os principais mecanismos de fotodegradação de moléculas orgânicas serão discutidos. Mecanismos de fotólise direta, e a utilização dos POAs em fotocatálise serão discutidos, sendo enfatizado o mecanismo de ação de semicondutores fotoexcitados. Desta forma, os embasamentos teóricos sobre o assunto, como apresentados, possibilitarão o melhor entendimento da relação existente entre os processos de síntese de fotocatalisadores nanoestruturados e suas implicações nas propriedades dos mesmos, como descrito nas seções posteriores.
Fotólise direta e POAs
Reações de foto-oxidação somente sob excitação eletrônica do substrato orgânico (molécula a qual se pretende degradar) são denominadas reações de fotólise direta e implicam, na maioria dos casos, em transferência eletrônica do substrato no estado excitado (C*. Equação 1) para uma molécula receptora, geralmente o oxigênio molecular em solução (Equação 2). Esta transferência eletrônica forma radicais catiônicos e aniônicos que podem sofrer subseqüente recombinação inibindo futuras reações, ou ainda pode ocorrer hidrólise do radical catiônico, responsável por posteriores reações de oxidação. O substrato pode também ser quebrado pela incidência da radiação (Equação 3) para formar radicais que reagem com o oxigênio molecular dissolvido (Equação 4) formando radicais orgânicos peroxil (RO2.). Estes radicais (RO2.) são considerados os verdadeiros propagadores da reação, podendo levar à completa mineralização do substrato.9

A fotólise direta, na maioria das vezes, não é capaz de promover a fotodegradação completa do substrato e geralmente provoca apenas algumas modificações na sua estrutura. No entanto, a sua utilização é bastante interessante quando a degradação por radical hidroxila (.OH) não é eficiente.9 A degradação de pesticidas, por exemplo, é possível através de diferentes processos fotoquímicos que necessitam de uma fonte de luz artificial ou luz solar natural, sendo que a maioria destes métodos requer períodos de longo tratamento com fótons de alta energia e raramente alcançam a completa degradação do poluente. Fontes de baixa energia (por exemplo, a radiação solar natural) não podem ser utilizadas eficientemente no tratamento da maioria dos resíduos, sendo necessária radiação com maior energia, especialmente na região do UVC.10 Assim, é notável que outros processos de fotodegradação precisem ser associados à fotólise direta. Alguns estudos comparativos demonstram essa necessidade de associação de processos. No entanto, demonstram também que, em algumas condições específicas,11,12 o processo de fotólise direta é mais eficiente que os processos indiretos.
Uma forma mais eficiente pra remoção de contaminantes é através de POAs.13 - 15 Estes baseiam-se na geração de radicais altamente reativos, tal como o radical hidroxila (.OH), um oxidante forte, não seletivo que reage com a maioria dos compostos orgânicos de forma muito rápida.Os radicais livres HO2.e O2.- também podem estar envolvidos nos processos de degradação, mas estes radicais são menos reativos do que os radicais hidroxila.7 A geração destes radicais pode ocorrer a partir de diversos processos, dos quais destacam-se UV/TiO2,16 - 20 UV/H2O2 e H2O2,21,22 Fenton e foto-Fenton,23,- 26 entre outros.27 - 29
O radical hidroxila (.OH) oxida compostos orgânicos principalmente pela abstração de hidrogênio (Equação 5). Essa reação gera radicais orgânicos que podem reagir com o oxigênio molecular para formar o radical peroxil (RO2.) (Equação 4), o qual inicia reações subsequentes de degradação oxidativa podendo formar subprodutos ou ser totalmente oxidado, dependendo das condições e substratos a serem degradados.9

Os radicais hidroxila (.OH) gerados na presença de um substrato orgânico, além da abstração de hidrogênio, podem também reagir por adição eletrofílica (Equação 6), transferência eletrônica (Equação 7). A recombinação radical-radical leva ao término da cadeia de reação (Equação 8).9

Fotocatálise com semicondutores
Dentre as maneiras de se gerar o radical responsável pelas reações de fotodegradação está o uso de semicondutores fotoativados. O processo global de fotodegradação de um composto orgânico utilizando-se semicondutores, quando ocorre a oxidação completa, pode ser representado pela seguinte equação reacional (Equação 9):30

Porém, em muitos processos de fotodegradação utilizando semicondutores ou outros fotocatalisadores, não se consegue chegar à oxidação completa do composto, observando-se somente a formação de subprodutos.
Semicondutores possuem potencial para serem utilizados como fotocatalisadores devido às suas estruturas eletrônicas serem caracterizadas por uma banda de valência (BV) preenchida e uma banda de condução (BC) vazia. Quando um fóton incide sobre a superfície de um semicondutor com energia hυ, igual ou maior à energia de band gap (Ebg) do semicondutor, um elétron (e-) é promovido da BV para BC, gerando um buraco (h+) na banda de valência. O elétron e o buraco podem recombinar-se, liberando a energia investida em forma de calor, ou reagir com aceptores e doadores de elétrons, respectivamente, que estejam adsorvidos na superfície do semicondutor.31 Na ausência de apropriados "sequestradores" para os buracos e elétrons, a energia armazenada é dissipada dentro de poucos nanosegundos pela recombinação destes. Caso um "sequestrador" consiga prender o elétron, a recombinação é prevenida e reações redox subsequentes podem ocorrer.31
O TiO2 na fase anatase demonstrou ser até momento o semicondutor mais adequado para utilização em fotocatálise devido à sua pequena taxa de recombinação.32As suas características espectrais de absorção - bandgap em 3,2 eV, correspondente a absorção óptica em 387 nm - permitem a sua excitação nas regiões de UVC (200-280 nm), UVB (280-320 nm), UVA (320-380 nm) e quando excitado eletronicamente exibe alto poder de oxidação devido ao buraco (h+) na banda de valência (Equação 10).9,30,31

Na banda de valência, duas reações de oxidação podem ser observadas: a transferência eletrônica direta do substrato orgânico (molécula) a ser degradado RX para o buraco (h+) (Equação 11)ou a transferência eletrônica para o buraco das espécies H2O e -OH adsorvidas (Equações 12 e 13) formando os radicais hidroxila (.OH). As reações de formação de .OH, aparentemente, são as de maior relevância no processo de degradação oxidativa, provavelmente devido à alta concentração de H2O e -OH adsorvidos na superfície do semicondutor.9,30,31

Na banda de condução, os elétrons deverão ser transferidos para um aceptor de elétrons. Como no processo de fotodegradação oxidativa o oxigênio molecular deverá estar presente, a transferência eletrônica geralmente ocorre da banda de condução do fotocatalisador para o oxigênio molecular dissolvido (Equação 14).9,30,31

Existem dois problemas principais para a aplicação em larga escala do TiO2 como fotocatalisador. O primeiro é que o TiO2 somente é ativado sob irradiação UV, mais especificamente radiação com comprimento de onda < 387 nm e, desta forma, não pode ser excitado utilizando-se radiação na região do visível. Assim, o uso da radiação solar é pouco eficiente no processo, visto que ela possui somente uma pequena fração (5%) de radiação UV. Procedimentos que possam estender a banda de absorção do TiO2 para a região do visível são, portanto, importantes para melhorar a eficiência fotocatalítica do TiO2, sob radiação solar. Para este fim, a modificação do TiO2 com dopantes não-metálicos, tais como C,33,34 N,35 e S,36,37 mostrou ser eficiente para promover a excitação do TiO2 sob luz visível e, consequentemente, radiação solar. Estes dopantes, quando incorporados à estrutura do TiO2, ampliam a banda de absorção deste material para comprimentos de onda maiores, ocasionando maior atividade fotocatalítica devido ao número maior de cargas fotogeradas.
O segundo problema é a recombinação das cargas fotogeradas (elétron e buraco) que diminui a eficiência do processo fotocatalítico. Desta maneira, aumentar a sobrevivência destas é fundamental para a fotodegradação mais eficiente. A incorporação de alguns metais (Ag, Pt, Pd, entre outros) à estrutura do semicondutor tem se mostrado eficiente no aumento da sua fotoatividade, através do aumento do tempo de vida das cargas fotogeradas. Anpo et al.38 reportaram maiores fotoatividades para sistemas TiO2/Pt em relação ao TiO2 puro. Wang et al.39 também reportaram o aumento da eficiência fotocatalítica de pós de TiO2 anatase de alta área superficial com a incorporação de nanopartículas de Ag. Este mesmo efeito aparece em junções de semicondutores ou suas soluções sólidas com diferentes valores de bandgap. Lin et al.40 reportaram maior atividade fotocatalítica de soluções sólidas de rutilo Ti1-xSnxO2 em relação ao TiO2 rutilo puro, na reação de oxidação de acetona. Vinodgopal et al.41 estudando a degradação de um corante demonstraram que filmes de compósitos SnO2/TiO2 apresentam velocidade de degradação maior do que filmes de SnO2 e TiO2 puros. Este aumento na velocidade de degradação do corante também foi atribuído a uma maior separação das cargas nestes compósitos, prevenindo a recombinação.
Em fotocatálise com semicondutores, a atividade fotocatalítica depende da excitação eletrônica do semicondutor e a consequente formação dos elétrons e buracos na banda de condução e valência, respectivamente. É notável que o poder de oxidação esteja relacionado com os buracos fotogerados e que a recombinação das cargas deva ser evitada para que as reações de fotodegradação ocorram. Com isso, boa parte dos trabalhos relacionados nesta área se concentra no aumento da atividade fotocatalítica destes sistemas pela diminuição da recombinação ou também pela ampliação da banda de absorção do fotocatalisador para regiões do espectro de mais baixa energia. Aliada a estas estratégias descritas anteriormente, a utilização de fotocatalisadores nanoestruturados é importante para o aumento da fotoatividade dos materiais, motivos estes já discutidos anteriormente neste trabalho. Conscientes da importância de nanoestruturas em fotocatálise foi feita uma ampla revisão sobre as principais metodologias de síntese de fotocatalisadores nanoestruturados e alguns exemplos de aplicações destes materiais.
MÉTODOS DE SÍNTESE DE NANOESTRUTURAS
Quando se trata de fotocatálise, é notável que o semicondutor mais utilizado seja o TiO2 na fase anatase. No entanto, atualmente vários trabalhos já reportam alta atividade fotocatalítica de ZnO,42 - 45 e também atividade fotocatalítica de CuO,46 ZnS,45 CdS,45 Ga2O3,47 entre outros.
O TiO2 é um óxido anfótero, embora mais ácido que básico, sendo também polimórfico, podendo existir em três formas cristalográficas: anatase (tetragonal), brookite (ortorrômbica) e rutilo (tetragonal).48 Brookite ocorre naturalmente, sendo extremamente difícil de sintetizar. Anatase e rutilo também ocorrem naturalmente, mas podem ser sintetizadas em laboratório. Porém, a obtenção da fase TiO2 anatase não é trivial, e constitui-se em um caso interessante do papel da escala nanométrica para a adequada obtenção do material.
Estudos experimentais e teóricos demonstram que a formação da fase anatase somente é preferencial em relação à fase TiO2 rutilo, de menor atividade fotocatalítica,49 para tamanhos inferiores a 12-13 nm considerando-se partículas esféricas, acima dos quais há a tendência de transformação de fase.50,51 Desta forma, metodologias de síntese do material capazes de controlar o tamanho de partícula final obtido são necessárias para desenvolvimento da fase.
Assim, esforços têm sido despendidos no intuito de se desenvolver métodos de síntese que possibilitem a obtenção de nanopartículas em uma determinada fase cristalina, com tamanho e morfologia controlada. Estes fatores são determinantes para que um material possua eficiência em uma determinada aplicação, tal como em fotocatálise. Desta maneira, os principais métodos utilizados em sínteses de nanoestruturas serão discutidos, destacando-se os métodos sol-gel hidrolítico e precursores poliméricos e, também, os métodos hidrotermais e solvotermais.
Método sol-gel hidrolítico
O termo "sol-gel" compreende diversos métodos de síntese, que se caracterizam por uma reação em meio homogêneo ("sol") que desestabilize o meio de forma controlada, formando partículas ou nanopartículas ("gel"). Essa definição ampla aplica-se a vários métodos, porém, na literatura, o termo é frequentemente associado ao processo hidrolítico, ou seja, a reação de um precursor anidro em meio aquoso. A rota de hidrólise envolve a ligação do íon OH- ao metal proveniente do precursor orgânico/haleto numa reação típica:52

onde M representa Si, Ti, Zr, Al e outros metais e R é um ligante orgânico. Um metal parcialmente hidrolisado pode reagir com outras moléculas parcialmente hidrolisadas numa reação de policondensação (Equações 16 e 17):

Este tipo de reação leva à formação de um polímero inorgânico, ou uma rede tridimensional formada por oxiânions metálicos.52,53 O processo permite boa homogeneidade e em geral leva à obtenção de fases metaestáveis, incluindo amorfas. Neste último caso (que é o mais comum) é necessária uma etapa de cristalização dos óxidos formados, termicamente por calcinação ou por tratamento hidrotermal. Vários fatores afetam o processo sol-gel, incluindo o tipo de precursor metálico, pH da solução, relação água/precursor, temperatura, natureza do solvente e estabilizantes.52 Variando-se estes parâmetros, as partículas podem ser sintetizadas com tamanho controlado, morfologia e estado de aglomeração. A hidrólise de haletos metálicos e outros sais inorgânicos é um método largamente empregado para obtenção de óxidos nanoparticulados, como TiO2,54 SnO2 puro e dopado,55,56 ZnO,57 ZrO2, Y2O3, entre outros. Na Figura 1 está apresentado um fluxograma descritivo da rota de síntese sol-gel com as principais etapas de síntese.
Método dos precursores poliméricos
O método dos precursores poliméricos é uma variante do processo sol-gel, no qual a reação de formação é não hidrolítica e gera um polímero, o qual, após calcinação ou outro método de eliminação do material orgânico, forma o óxido de interesse.60,61 O processo é uma alternativa viável para sínteses com um rigoroso controle da estequiometria, morfologia do pó e pureza de fase. Este método consiste primeiramente na formação de um complexo entre um ácido hidroxicarboxílico (usualmente o ácido cítrico) e cátions dissolvidos como sais em uma solução aquosa. O complexo formado é misturado a um poliálcool (geralmente etilenoglicol) sendo levemente aquecido (80-110 ºC) até a obtenção de uma solução geralmente transparente. Um novo aquecimento (150-250 ºC) causa a reação de condensação com formação de uma molécula de água e se o aquecimento persistir, acontece a poliesterificação e o excesso de água é removido resultando em uma resina polimérica sólida.62
A idéia central do método é distribuir os cátions por toda a estrutura polimérica. Um tratamento térmico em temperaturas mais altas (a partir de 400 ºC) causa a liberação da matéria orgânica e a formação dos cristalitos devidamente ordenados. Este resultado é particularmente interessante quando se deseja a obtenção de materiais com alta cristalinidade e controlada distribuição dos constituintes na rede cristalina. A principal desvantagem deste método é o fato de que o aquecimento necessário para liberação de toda matéria orgânica pode ocasionar o crescimento das partículas, contudo este parâmetro pode ser satisfatoriamente controlado pela relação metal:orgânicos. Diversos trabalhos reportaram a síntese de materiais nanoestruturados, utilizando-se o método dos precursores poliméricos, incluindo a síntese de nanopartículas de TiO2,63 titanatos,64 - 66 nanocompósitos,67 filmes nanoestruturados de TiO2,68 de SnO2 puro e dopado,69 entre outros. Na Figura 2 está apresentado o fluxograma com as principais etapas de síntese do método dos precursores poliméricos.
Método hidrotérmico
Uma alternativa para cristalização sem o uso de tratamento térmico em temperaturas elevadas é o uso de condições hidrotermais e solvotermais. Em condições hidrotermais a solubilidade das partículas amorfas é significantemente aumentada e a cristalização pode ocorrer concomitantemente com processos de redissolução e reprecipitação - porém no núcleo cristalino. Também, o aumento da solubilidade das partículas favorece processos de crescimento por Ostwald ripening,70,- 72 o que torna a estratégia útil para estas observações. Trabalhos recentesdemonstraram também a ocorrência do fenômeno de coalescência orientada em partículas hidrotermalizadas, provavelmente por aumento de eventos de colisão interpartículas, mostrando que este fenômeno também pode ser considerado neste processo.73 - 78
A Figura 3 apresenta o esquema do aparato normalmente utilizado em ensaios hidrotermais. Consiste basicamente de um reator em aço inox com uma cápsula interna de politetrafluoretileno (PTFE), para minimização de efeitos de ataque químico por parte da solução, o qual é aquecido externamente por um forno tubular. Uma sonda interna de temperatura e uma sonda de pressão mantêm o controle do sistema, que pode assim operar em temperatura constante, a uma pressão de até 40 atm, por longos períodos de tempo.
O tratamento hidrotermal já foi muito utilizado na síntese de nanoestruturas de semicondutores. Pequenas variações nos principais parâmetros de síntese (pH, concentração do precursor, entre outros) podem causar substanciais mudanças na morfologia, tamanho, constituição química, entre outras propriedades das nanoestruturas sintetizadas.
Desta forma, alguns trabalhos reportaram a síntese de TiO2 através de tratamento hidrotermal, sob várias temperaturas e valores de pH.79,80 Para estes estudos, efetuou-se o tratamento hidrotermal dos pós de TiO2 amorfos previamente obtidos, e o controle do pH foi feito com soluções de HNO3 ou KOH, escolhidos devido ao fato de seus contra-íons não se intercalarem na rede cristalina do produto cristalino final. Pôde-se observar que TiO2 na fase rutilo é obtido unicamente em baixos valores de pH e, consequentemente, o acréscimo no valor do pH favorece a formação da fase anatase.
A concentração do precursor no reator hidrotermal possui um papel importante na fase e morfologia obtida. Isto acontece, pois a concentração é um fator chave em uma determinada cinética reacional, influenciando na mobilidade das partículas suspensas e na proporção de choques efetivos. Em um estudo detalhado, utilizando o tratamento hidrotermal de solução de TiCl4 foi possível explicar a relação entre tamanho de cristalito e a concentração do reagente por meio de uma relação matemática semiquantitativa obtida a partir da teoria de nucleação e crescimento.81
Como o processo fotocatalítico é um fenômeno de superfície, a análise dos planos cristalográficos preferencialmente expostos numa determinada morfologia se torna importante. Superfícies sólidas de diferentes orientações cristalográficas possuem diferentes energias de superfície e, consequentemente, diferentes afinidades com determinados íons ou moléculas. Nesse contexto, foi desenvolvido um processo de síntese hidrotermal para o TiO2 anatase em ambiente contendo íons fluoretos.82 Este processo proporciona cristais com alta porcentagem de planos {001}, que são as facetas mais reativas destes cristais.83 Estes íons têm a capacidade de quando incorporados nesta superfície, em substituição aos átomos de O, tornar os planos {001} mais estáveis em relação aos planos {101}, estes reportados como de menores energias,83 induzindo o crescimento na direção que resulta em menor energia superficial para partícula final. A maior estabilidade dos planos onde os íons fluoretos estão incorporados deve-se ao fato de que a ligação F-Ti possui alta energia. Ao final da reação, o produto pode ser purificado pelo simples aquecimento a 600 ºC, sem mudanças na estrutura cristalina ou na morfologia do material obtido.
Com o objetivo de se induzir o crescimento em uma determinada direção, também é comum a utilização de surfactantes que interajam preferencialmente com determinada família de planos. Neste caso, a direção de crescimento é ditada tanto pela energia superficial quanto pelo efeito estérico gerado pelo surfactante adsorvido. Alivisatos et al.84 reportaram a obtenção de cristais, em morfologias que vão desde nanoesferas a nanofios, em meio não aquoso, com condições de síntese constante, unicamente por tratamentos com surfactantes.
Han et al.85 reportaram a síntese de TiO2, em baixas temperaturas, apenas variando-se o pH e os íons presentes no ambiente de síntese. Os testes fotocatalíticos com essas amostras mostraram a influência da morfologia na eficiência fotocatalítica do material, sendo este resultado justificado em termos da área superficial específica de cada morfologia. Em processos de síntese semelhantes aos descritos acima, o aumento do pH para valores extremamente básicos tende a resultar na formação de nanoestruturas de Ti com características tubulares. A Figura 4 apresenta imagens de microscopia eletrônica de varredura (MEV) que ilustram a influência do valor do pH na síntese de nanoestruturas de TiO2. Pode-se observar que o aumento do pH resulta na formação de nanoestruturas mais alongadas. Em condições altamente alcalinas, o material obtido apresenta características tubulares.80
Estruturas tubulares em nanoescala são interessantes devido à sua alta área superficial específica que é um fator decisivo em fotocatálise. Desde 1991, quando Iijima86 reportou pela primeira vez a síntese de nanotubos de carbono, têm surgido diversas pesquisas de síntese de nanotubos, inclusive de semicondutores. Kasuga et al.87 reportaram a síntese de cristais de TiO2 anatase em forma de agulha (nanoestruturas tubulares) através do tratamento hidrotérmico de TiO2 com solução aquosa de NaOH. Foram avaliados os efeitos da variação da concentração da base e temperatura de tratamento térmico sobre a morfologia das nanoestruturas formadas. Foram obtidas nanoestruturas tubulares com aproximadamente 8 nm de diâmetro, 100 nm de comprimento e alta área superficial de 400 m2 g-1, sob tratamento térmico de TiO2 com solução aquosa de NaOH 10 M a 110 ºC por 20 h. A avaliação da influência das principais variáveis de síntese revelou que soluções diluídas de NaOH e baixas temperaturas de tratamento possuem pouca habilidade em formar nanoestruturas tubulares. Morgan et al.88 também estudaram variáveis de síntese hidrotermal utilizando TiO2 comercial (Degussa P25) como precursor. Foi investigado o efeito da concentração da base e da temperatura de tratamento hidrotermal. De acordo com os resultados, como já observado por Kasuga et al., as condições de mais alta temperatura e concentração da base são mais favoráveis à formação de nanoestruturas tubulares.
Kasuga também reportou a síntese de cristais de TiO2 com características semelhantes às citadas no trabalho anterior.89 As nanoestruturas foram sintetizadas através do tratamento hidrotermal com solução aquosa de NaOH 10 M a 110 ºC por 20 h e posteriormente lavagem com solução aquosa de HCl. Imagens obtidas por microscopia eletrônica de transmissão mostraram nanoestruturas tubulares com diâmetros interno e externo de 5 e 8 nm, respectivamente. A área superficial específica da amostra de TiO2 antes do tratamento químico era de 50 m2 g-1, aumentando para 400 m2 g-1 após o tratamento. O estudo da composição química dos nanotubos demonstrou que estes não seriam formados exclusivamente de titânia (TiO2) e sim uma mistura de titânia e titanatos.
Du et al.90 também reportaram a síntese de nanomateriais tubulares utilizando procedimento semelhante ao descrito por Kasuga et al.. Cristais de TiO2 compostos pelas as fases anatase e rutilo foram transformados em nanoestruturas tubulares com alta cristalinidade. Foram identificadas nanoestruturas tubulares antes e após o tratamento com HCl, sendo estas compostas por múltiplas "cascas"com 8-10 nm de diâmetro e comprimentos variando entre várias dezenas a várias centenas de nanômetros. Análises por EDS - energy dispersion spectroscopy - indicaram que os nanotubos eram compostos basicamente por Ti e O. Adicionalmente, o estudo da composição química dos nanotubos demonstrou que eram formados por camadas compostas somente por titanatos, do tipo H2Ti3O7.
Ma et al.91 reportaram a síntese de nanotubos de titanato também através do tratamento hidrotermal de TiO2 em solução de NaOH. Foram formados nanotubos com diâmetro interno de 5 nm e externo de 10 nm, sendo compostos por três a cinco camadas de titanatos. Estruturas tubulares foram evidenciadas mesmo antes da lavagem com HCl e foram apresentadas fortes evidências, através de DRX e observações microscópicas de alta resolução, de que os nanotubos são constituídos de lâminas de Lepidocrocitas HxTi2-x/4x/4O4 (x~0,7 e = vacância). Procedimentos semelhantes foram utilizados em outros trabalhos para a síntese de nanoestruturas tubulares de titanatos e/ou titânia utilizando tratamento hidrotermal alcalino de pós de TiO2.92 ,- 97
Trabalhos recentes reportaram também uma maior fotoatividade para nanoestruturas tubulares dopadas com materiais que ampliam a banda de absorção para a região do visível. Geng et al.98 reportaram a síntese de nanotubos de titanato puro e dopado com carbono, através de tratamento hidrotermal. Para obtenção dos nanotubos dopados com carbono, os nanotubos puros foram dispersos em solução aquosa de hidróxido de tetraetilamônio e, posteriormente, foi feita a calcinação desta suspensão, obtendo-se assim os nanotubos de titanato dopados com carbono. Os nanotubos dopados exibiram maior fotoatividade, sob radiação solar simulada, para a degradação de azul de metileno do que os nanotubos não modificados. Resultados semelhantes foram obtidos dopando-se os nanotubos de titanato com nitrogênio.99
A dopagem de nanoestruturas tubulares com metaismostrou-se eficiente para prevenir a recombinação. Zhu et al.100 reportaram a preparação por tratamento hidrotermal de nanoestruturas tubulares de TiO2/Pd com grande performance fotocatalítica. Han et al.101 reportaram a síntese de nanotubos de titanato/Pd ou Pt por tratamento hidrotermal com alta eficiência como sensores de gás hidrogênio. Pó de titânia anatase comercial e PdCl2 ou H2PtCl6 em uma quantidade igual foram dispersos a 40 mL de uma solução aquosa de NaOH 10 M e colocados sob tratamento hidrotermal a 150 ºC por 12 h. Os precipitados foram separados por filtração e lavados com HCl diluído e água deionizada. Posteriormente, os nanotubos de Titanato/Pd e Pt foram secos a 120 ºC.
Método solvotérmico
Uma variante do método hidrotérmico é a reação em meio não aquoso, normalmente chamada de método solvotérmico. Nestas condições, reações não hidrolíticas podem ser favorecidas, permitindo maior controle de etapas de reação pelo impedimento estérico das cadeias orgânicas envolvidas. O método foi muito explorado para reações de precursores metálicos em álcool benzílico, um solvente que se demonstrou hábil para atuar como reagente e surfactante das partículas formadas.102-105De forma geral, o método consiste na dissolução de um precursor metálico em álcool benzílico anidro e solvotermalização em temperaturas superiores a 150 ºC. Nestas condições, a hidroxila presente no álcool liga-se parcialmente ao íon metálico, iniciando uma reação de policondensação, na qual o grupo orgânico é participante. O impedimento estérico exercido pelo maior volume do grupo orgânico age controlando a formação das nanopartículas, que tendem a se estabilizar em tamanhos menores e em dispersões de tamanhos mais estreitas que as obtidas no processo sol-gel hidrolítico.104 Vários óxidos semicondutores foram obtidos pela estratégia, como óxido de índio dopado com estanho (ITO),106,107 ZrO2,108 V2O3,109 Nb2O5,110 e TiO2 anatase.111,112
A estratégia, apesar de demonstrar vantagens no controle da síntese, apresenta como principal problema a possibilidade da presença de grupos orgânicos remanescentes na superfície das nanopartículas formadas, o que poderia envenenar sítios catalíticos. Porém, estudos recentes têm demonstrado que é possível remover os grupos orgânicos, trocando-os por grupos hidrofílicos e redispersando-os em outros solventes.108
Métodos de síntese solvotermais com outros solventes não reativos são também explorados no sentido de se obter nanopartículas de alta cristalinidade. Perera et al.113 obtiveram nanocristais de anatase e mistura de fase anatase/brookite através do tratamento de TiBr4 em hexadecano, na presença de Na2O2. Esse solvente foi utilizado devido ao seu alto ponto de ebulição, e a diferença no produto final se deve a diferentes taxas de aquecimento empregadas durante a síntese. O cristal obtido apresentou maior eficiência quando testado frente à fotodegradação do azul de metileno, em relação ao TiO2 comercial Degussa P-25. Este resultado deve-se, provavelmente, à maior área superficial do nanocristal sintetizado via método solvotermal em relação ao TiO2 comercial. Mesmo a mistura de fase anatase/brookite apresentou maior eficiência na fotodegradação do composto orgânico, porém neste caso é provável a existência de efeitos de interface entre fases, que facilita a separação entre as cargas fotogeradas no processo.114Interessante notar que a fase brookite é relegada como fase não ativa em fotocatálise, sendo indesejada em muitos processos de síntese.115
Qin et al.116 reportaram a síntese de nanocristais de TiO2 em suas três estruturas cristalinas, dependendo do solvente utilizado. A fase rutilo foi obtida em metanol sob condições básicas, indo de encontro a relatos na literatura,115 onde esta fase foi apenas obtida em valores de pH extremamente ácidos, porém em ambiente aquoso. Testes fotocatalíticos novamente mostraram uma maior eficiência quando se observa a mistura de fases dos polimorfos do material.
Relatos de controle morfológico em nanocristais de TiO2 sintetizados via método solvotermal são também comuns. Xie et al.117 reportaram este procedimento introduzindo etilenodiamina (EDA) em diferentes concentrações em um sistema simples formado por isopropóxido de titânio e etilenoglicol. Neste ambiente de síntese, o EDA atua como inibidor do crescimento, uma vez que na sua ausência foram obtidos nanofios, e após sua introdução no ambiente de síntese e dependendo da sua concentração, obtiveram-se nanofios ou até emaranhados de nanofibras com dimensões menores que os produtos obtidos na ausência do EDA.
ESTRATÉGIA DE APLICAÇÃO DE NANOESTRUTURAS EM FOTOCATÁLISE
Nanocompósitos
Certamente, a estratégia mais simples de utilização de semicondutores em fotocatálise é como suspensão coloidal na solução contendo a molécula que se pretende degradar. Uma característica das suspensões coloidais de óxidos simples nanoestruturados, como TiO2, é sua alta estabilidade em meio aquoso.118 Porém, esta característica torna-se um complicador ao fim do processo de fotodegradação, visto que a estabilidade do colóide dificulta a separação das mesmas nanopartículas para reutilização. Desta forma, o processo de estabilização-desestabilização do colóide pode ser visto como um dos principais obstáculos ao uso da metodologia em larga escala.
Uma forma simples de separação é associar às nanopartículas de interesse uma fase com atividade magnética como, por exemplo, ferritas (CoFe2O4 ou Fe3O4 (magnetita)), através da formação de um nanocompósito, o qual pode ser submetido à ação de um campo magnético. Beydoun et al.119,120 reportaram a síntese de magnetita Fe3O4 recoberta com TiO2 (TiO2/Fe3O4) utilizando a técnica sol-gel no processo de recobrimento. O método utilizado pelos autores mostrou-se eficiente na formação das heteroestruturas, visto que o material obtido apresentou atividade fotocatalítica associada à propriedade magnética. Porém, os autores reportaram redução da atividade fotocatalítica dos nanocompósitos comparativamente ao TiO2 puro, associando o fato a mecanismos de recombinação na interface das fases.
A efetividade do arraste magnético como estratégia de separação de nanopartículas pode ser visto em outros trabalhos. Lin et al.121 reportaram a formação de nanopartículas da ordem de 50 nm de magnetita encapsuladas com sílica, com boa dispersão das nanopartículas em água na ausência de campo magnético e com efetiva captura magnética das mesmas. As nanopartículas encapsuladas foram obtidas pela dispersão de nanopartículas de Fe3O4 em solvente, procedendo na dispersão à hidrólise do tetraetilortossilicato (TEOS), precursor da sílica. A formação de sílica ocorreu na superfície das nanopartículas superparamagnéticas, resultando no encapsulamento desejado. Os autores demonstraram a efetividade da estratégia através da adsorção de um corante à superfície das nanopartículas em suspensão, modificando sua coloração e eliminando seletivamente o mesmo corante através do arraste magnético das nanopartículas.
A literatura descreve várias outras estratégias de recobrimento, que podem ser utilizadas para o mesmo fim. Maciel et al.122 demonstraram a possibilidade de uso do método dos precursores poliméricos para obtenção de aluminas recobertas com óxido de lantânio, formando camadas homogêneas de espessuras da ordem de 5-10 nm. A mesma rota foi empregada por Libanori et al.123 para obtenção de nanopartículas de TiO2 modificadas superficialmente com Y2O3 e Al3O3. Ribeiro e Mourão também reportaram a síntese de nanocompósitos de CoFe2O4@TiO2 através do método dos precursores poliméricos.67 Os resultados obtidos demonstraram que o método dos precursores poliméricos é viável como alternativa para obtenção de nanocompósitos de CoFe2O4@TiO2, com potencial para uso como fotocatalisadores da degradação de pesticidas, através da avaliação da fotodegradação do herbicida atrazina. A Figura 5 apresenta uma imagem dos nanocompósitos magnéticos dispersos em meio aquoso e submetidos a um campo
Filmes nanoestruturados
Como já citado, o uso de fotocatalisadores nanoestruturados em suspensão possui como grande deficiência a dificuldade de separação do material após o processo de fotodegradação. Além da utilização de nanocompósitos com atividade magnética, a utilização do semicondutor imobilizado, especificamente na forma de filmes finos, é uma boa alternativa para contornar este problema. Além desta vantagem, a alta estabilidade mecânica dos filmes é outra ótima característica da utilização de sistemas imobilizados. No entanto, deve-se ressaltar que a utilização de filmes em fotocatálise pode diminuir a eficiência do processo devido à diminuição da área superficial em relação ao material particulado, ou pode dificultar a elaboração de fotorreatores para o tratamento de grandes volumes de resíduos.
Na literatura, vários trabalhos têm surgido sobre a preparação de filmes finos de TiO2 por diferentes técnicas químicas e não químicas, tais como pirólise,124magnetron sputtering,125 aquecimento por radiação microondas,126spin-coating,127dip-coating,128 hidrólise térmica,129 eletrodeposição catódica,130 métodos eletroquímicos,131 entre outras.
No processo de deposição por solução química, podem ser utilizadas várias técnicas de deposição, entre elas o spin-coating e dip-coating. A técnica spin-coating consiste em gotejar a solução precursora ou suspensão de nanopartículas sobre um substrato e o submeter a uma rotação controlada de forma a espalhar homogeneamente a solução/suspensão sobre o substrato.132 - 135 Nesse processo, são controlados a velocidade e o tempo de deposição da resina. Já a técnica de dip-coating consiste de um motor de passo que, ao movimentar a alavanca em que está fixado o substrato, permite imergir e retirar o substrato da solução em velocidade lenta e constante para que a solução possa aderir ao substrato.136,137
Diversos trabalhos têm surgido sobre a utilização de métodos químicos para a obtenção de filmes finos fotocatalisadores. No trabalho de Li et al.138 foi utilizado um método químico para preparar filmes fotocatalisadores porosos de CdIn2S4 altamente cristalinos a baixa temperatura sobre substrato de vidro. A utilização do método sol-gel com a técnica de spin-coating para preparar filmes finos consistindo de um compósito nanocristalino de TiO2/Ag foi reportada no trabalho de Chang et al.139 Os autores investigaram as propriedades fotocatalíticas destes filmes através da degradação do azul de metileno sob irradiação UV. Sayılkan et al.140 sintetizaram filmes finos nanoestruturados de TiO2 dopados com Sn e não dopados, pelo processo hidrotermal a 225 ºC durante 1 h. Neste trabalho, também foram preparados filmes finos fotocatalisadores baseados em nanoestruturas de TiO2, contendo diferentes razões sólidas de TiO2 na deposição sobre superfícies de vidro utilizando a técnica de spin coating.
Malagutti et al.68 sintetizaram filmes nanoestruturados de TiO2 a partir de resinas preparadas pelo método dos precursores poliméricos depositadas sobre substratos de vidro. A Figura 6 apresenta uma imagem de microscopia de força atômica (MFA) do filme. Foram obtidos filmes contínuos e regulares, sem separação de fase, demonstrando que o método de síntese utilizado resultou na formação de filmes nanoestruturados com alta homogeneidade, como pode ser observado na figura.
Negishi et al.141 reportaram a síntese de filmes finos de TiO2 depositados sobre vidro através do processo de dip coating, utilizando-se um solvente altamente viscoso. Dois tipos de procedimentos de preparação foram utilizados para as amostras, um sistema sol-gel e um sistema de decomposição térmica. A atividade fotocatalítica dos filmes preparados foi avaliada frente à foto-oxidação do NO e foi observado que os sintetizados por sol-gel e por decomposição térmica apresentaram atividade fotocatalítica semelhante, indicando que os dois métodos possuem grande potencial para obtenção de filmes finos de TiO2 ativos. Hsu et al.142 sintetizaram filmes finos de nanotubos de TiO2 dopados com nitrogênio (N-TNT) utilizando o método sol-gel e a técnica de dip-coating. Yuan et al.143 também utilizaram a técnica para depositar filmes.
Quando se trata de filmes finos aplicados em fotocatálise alguns parâmetros de síntese, tais como o método de síntese e deposição utilizados, temperatura e tempo de tratamento, podem interferir na cristalinidade, fase cristalina, tamanho dos grãos, área superficial fotocataliticamente ativa, entre outras características destes filmes.
Assim, a preparação de filmes finos de TiO2 à baixa temperatura utilizando sol-gel foi reportada por Biswas et al..128 Os filmes finos foram preparados sobre substrato de vidro pelo método sol-gel utilizando dip-coating como técnica de deposição, sendo avaliado o efeito da temperatura de tratamento na cristalinidade e atividade fotocatalítica dos filmes frente à decomposição de metanol, sob irradiação UV-vis. Os autores mostraram que a velocidade de decomposição do álcool depende das propriedades estruturais e superficiais dos filmes de TiO2, sendo que os filmes preparados em temperaturas menores apresentaram maior atividade fotocatalítica. Este efeito deve-se à alta porosidade, com grãos pequenos e a superfície aberta dos filmes que foram submetidos a temperaturas mais brandas. Através dos resultados pode-se sugerir que apesar de fotocatalisadores com maior cristalinidade possuírem a princípio maior potencial para aplicações fotocatalíticas, a perda de porosidade e área superficial (devido ao crescimento das partículas) são negativas para a eficiência dos filmes nos processos fotocatalíticos.
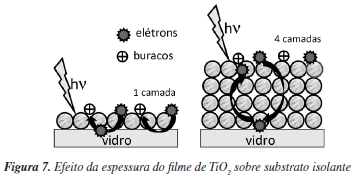
A espessura da camada depositada do filme também é um fator que pode influenciar a fotoatividade dos filmes. Por exemplo, para filmes de TiO2 depositados sobre suportes de vidro (substrato isolante), o aumento da espessura pode ser favorável para a eficiência destas estruturas.68 Isto pode ser explicado pelo fato de que filmes menos espessos possuem maior velocidade de recombinação das cargas do que filmes com maior espessura. A Figura 7 ilustra este efeito, onde se observa que o elétron fotogerado sobre a superfície do filme com apenas uma camada de deposição (lado esquerdo), percorre uma distância menor entre a sua fotogeração na superfície, colisão com o substrato isolante e subsequente recombinação com o buraco na superfície, do que o elétron fotogerado no filme de quatro camadas. É notável, porém, que estes efeitos foram observados para filmes nanoestruturados de TiO2 puro sobre suporte de vidro e deve-se considerar a possibilidade de que outros mecanismos possam existir para filmes com outras peculiaridades, tais como: substratos condutores, outros materiais depositados (como por exemplo, TiO2 dopado) e camadas de deposição mais espessas que as estudadas.
Além dos métodos químicos para a preparação de filmes finos mencionados anteriormente, também se podem citar outros métodos como, por exemplo, no trabalho de Kitano et. al.,144 que reporta a preparação de filmes finos fotocatalisadores de TiO2 com nitrogênio substituído (N-TiO2) utilizando o método de deposição de radio frequency magnetron sputtering (RF-MS). Yamaki et al.145 reportaram a preparação de filmes finos de TiO2 sobre um substrato de safira utilizando deposição com laser pulsado (PLD). Quan et al.146 sintetizaram eletrodos de filme de nanotubos de TiO2 utilizando o método de oxidação anódica. Subramanian et al.147 utilizaram um sistema eletroforético para depositar partículas de metais nobres de Au, Pt, e Ir sobre filmes nanoestruturados de TiO2. Outros trabalhos também reportaram a síntese filmes nanoestruturados.148 - 151
CONSIDERAÇÕES FINAIS
Existem enormes dificuldades que podem ser encontradas na instalação de sistemas fotocatalíticos reais operando em grande escala, incluindo aspectos econômicos, separação do fotocatalisador após o processo de descontaminação, adequação do sistema estudado em laboratório para as necessidades específicas de cada resíduo e, principalmente, a baixa eficiência de alguns métodos. Quando se utilizam semicondutores como fotocatalisadores, o aumento da área superficial através da utilização de nanoestruturas é altamente favorável à efetividade destas estruturas no processo. O aumento da efetividade destes processos com a utilização de nanoestruturas pode colaborar para o fortalecimento do uso dos POAs em alguns setores produtivos, bem como trabalhos sobre metodologias de aplicação - formação de nanocompósitos ou imobilização - serão fundamentais para viabilização das tecnologias. Assim, este trabalho apresenta uma ampla revisão sobre metodologias de síntese de nanoestruturas com potencial para serem utilizadas como fotocatalisadores e permitirá uma melhor disseminação destas estratégias de síntese de nanoestruturas e, consequentemente, do uso destas em processo fotocatalíticos.
AGRADECIMENTOS
À FAPESP, ao CNPq e FINEP pelo suporte financeiro.
Recebido em 19/11/08; aceito em 29/4/09; publicado na web em 6/10/09
- 1. Almeida, E.; Assalini, M. R.; Rosa, M. A.; Durán, N.; Quim. Nova 2004, 27, 818.
- 2. Lagaly, G.; Appl. Clay Sci 2001, 18, 205.
- 3. Cordeiro, A. C. de S.; Leite, S. G. F.; Dezotti, M.; Quim. Nova 2004, 27, 689.
- 4. Kondo, M. M.; Orlanda, J. F. F.; Ferreira, M. G. A. B.; Grassi, M. T.; Quim. Nova 2003, 26, 133.
- 5. Morais, J. L.; Sirtori, C.; Peralta-Zamora, P. G.; Quim. Nova 2006, 29, 20.
-
6Brasil, Diário Oficial da União- Nº 5, 8 de janeiro de 2008.
- 7. Chiron, S.; Fernandez-Alba, A.; Rodriguez, A.; Garcia-Calvo, E.; Water Res 2000, 34, 366.
- 8. Boudart, M.; Djega-Mariadasson, G.; Kinetics of Heterogeneous Catalytic Reactions, Princeton University Press: Princeton, 1984, 1Ş ed.
- 9. Legrini, O.; Oliveros, E.; Braun, A. M.; Chem. Rev. (Washington DC, U. S.) 1993, 93, 671.
- 10. Teixeira, S. C. G.; Canela, M. C.; Quim. Nova 2007, 30, 1830.
- 11. Garbin, J. R.; Milori, D. M. B. P.; Simões, M. L.; da Silva, W. T. L.; Martin-Neto, L.; Chemosphere 2007, 66, 1692.
- 12. Macounová, K.; Urban, J.; Krýsová, H.; Krýsa, J.; Jirkovský, J.; Ludvík, J.; J. Photochem. Photobiol., A 2001, 140, 93.
- 13. Nogueira, R. F.P.; Jardim, W. F.; Quim. Nova 1998, 21, 69.
- 14. Nogueira, R. F. P.; Jardim, W. F.; Sol. Energy Mater. Sol. Cells 1996, 56, 471.
- 15. Moctezuma, E.; Leyva, E.; Monreal, E.; Villegas, N.; Infante, D.; Chemosphere 1999, 32, 511.
- 16. Ziolli, R. L.; Jardim, W. F.; Quim. Nova 1998, 21, 319.
- 17. Pascoal, S. de A.; Lima, C. A. P.; Sousa, J. T.; Lima, G. G. C.; Vieira, F. F.; Quim. Nova 2007, 30, 1082.
- 18. Mills, A.; Hill, G.; Crow, M.; Hodgen, S.; J. Appl. Electrochem. 2005, 35, 641.
- 19. Wu, G.; Chen, A.; J. Photochem. Photobiol., A 2008, 195, 47.
- 20. Osugi, M. E.; Umbuzeiro, G. A.; Anderson, M. A.; Zanoni, M. V. B.; Electrochim. Acta 2005, 50, 5261.
- 21. Araujo, F. V. F.; Yokoyama, L.; Teixeira, L. A. C.; Quim. Nova 2006, 29, 11.
- 22. Gromboni, C. F.; Ferreira, A. G.; Kamogawa, M. Y.; Nogueira, A. R. de A.; Quim. Nova 2007, 30, 264.
- 23. Carneiro, P. A.; Nogueira, R. F. P.; Zanoni, M. V. B.; Dyes Pigm. 2007, 74, 127.
- 24. Britto, J. M.; Rangel, M. do C.; Quim. Nova 2008, 31, 114.
- 25. Tiburtius, E. R. L.; Peralta-Zamora, P.; Emmel, A.; Leal, E. S.; Quim. Nova 2005, 28, 61.
- 26. Pignatello, J. J.; Environ. Sci. Technol. 1992, 26, 944.
- 27. Almeida, E.; Assalin, M. R.; Rosa, M. A.; Duran, N.; Quim.Nova 2004, 27, 818.
- 28. Pereira, W. S.; Freire, R. S.; Quim. Nova 2005, 28, 130.
- 29. Mahmoud, A.; Freire, R. S.; Quim. Nova 2007, 30, 198.
- 30. Mills, A.; Davies, R. H.; Worsley, D.; Chem. Soc. Rev. 1993, 22, 417.
- 31. Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W.; Chem. Rev.(Washington DC, U. S.) 1995, 95, 69.
- 32.Linsebigler, A. L.; Lu, G.; Yates, J. T.; Chem. Rev. (Washington DC, U. S.) 1995, 95, 735.
- 33. Khan, S. U. M.; Al-Shahry, M.; Ingler Jr., W. B.; Science (Washington DC, U. S.) 2002, 297, 2243.
- 34. Irie, H.; Watanabe, Y.; Hashimoto, K.; Chem. Lett 2003, 32, 772.
- 35. Burda, C.; Lou, Y.; Chen, X.; Samia, A. C. S.; Stout, J.; Gole, J. L.; Nano Lett. 2003, 3, 1049.
- 36. Umebayashi, T.; Yamaki, T.; Yamamot, S.; Tanaka, S.; Appl. Phys. Lett 2003, 93, 5156.
- 37. Ohno, T.; Mitsui, T.; Matsumura, M.; Chem. Lett 2003, 32, 364.
- 38. Anpo, M.; Takeuchi, M.; J. Catal. 2003, 216, 505.
- 39. Wang, H. W.; Lin, H. C.; Kuo, C. H.; Cheng, Y. L.; Yeh, Y. C.; J. Phys. Chem Solids 2008, 69, 633.
- 40. Lin, J.; Yu, J. C.; Lo, D.; Lam, S. K.; J. Catal. 1999, 183, 368.
- 41. Vinodgopal, K.; Bedja, I.; Kamat, P. V.; Chem. Mater. 1996, 8, 2180.
- 42. Kansal, S. K.; Singh, M.; Sud, D.; J. Hazard. Mater. 2008, 153, 412.
- 43. Hariharan, C.; Appl. Catal., A 2006, 304, 55.
- 44. Jung, H.; Choi, H.; Appl. Catal., B 2006, 66, 288.
- 45. Kansal, S. K.; Singh, M.; Sud, D.; J. Hazard. Mater. 2007, 141, 581.
- 46. Ben-Moshe, T.; Dror, I.; Berkowitz, B.; Appl. Catal., B 2009, 85, 207.
- 47. Srihari, V.; Sridharan, V.; Sahu, H. K.; Raghavan, G.; Sastry, V. S.; Sundar, C. S.; J. Mater. Sci. 2009, 44, 671.
- 48. Gribb, A. A.; Banfield, J. F.; Am. Mineral. 1997, 82, 717.
- 49. Sun, B.; Vorontsov, A. V.; Smirniotis, P. G.; Langmuir 2003, 19, 3151.
- 50. Barnard, A. S.; Zapol, P.; J. Phys. Chem. B 2004, 108, 18435.
- 51. Barnard, A. S.; Zapol, P.; Phys. Rev. B 2004, 70, 235403.
- 52. Brinker, C. J.; Scherrer, G. W.; Sol-Gel Science, Academic Press: Boston, 1990, 1Ş ed.
- 53. Ying, J. Y.; Chem. Mater. 1997, 9, 2247.
- 54. Ragai, J.; Lotfi, W.; Colloids Surf. 1991, 61, 97.
- 55. Nutz, T.; Haase, M.; J. Phys. Chem. B 2000, 104, 8430.
- 56. Ribeiro, C.; Lee, E. J. H.; Giraldi, T. R.; Varela, J. A.; Longo, E.; Leite, E. R.; J. Phys. Chem. B 2004, 108, 15612.
- 57. Meulenkamp, E. A.; J. Phys. Chem. B 1998, 102, 7764.
- 58. Hu, M. Z. C.; Hunt, R. D.; Payzant, E. A.; Hubbard, C. R.; J. Am. Ceram. Soc. 1999, 82, 2313.
- 59. Fokema, M. D.; Chiu, E.; Ying, J. Y.; Langmuir 2000, 16, 3154.
- 60. Kakihana, M.; Yoshimura, M.; Bull. Chem. Soc. Jpn. 1999, 72, 1427.
- 61. Arima, M.; Kakihana, M.; Nakamura, Y.; Yashima, M.; Yoshimura, M.; J. Am. Ceram. Soc. 1996, 79, 2847.
- 62. Lessing, P. A.; Am. Ceram. Soc. Bull. 1989, 68, 1002.
- 63. Ronconi, C. M.; Ribeiro, C.; Bulhões, L. O. S.; Pereira, E. C.; J. Alloys Compd. 2008, 466, 435.
- 64. Kakihana, M.; Okubo, T.; Arima, M.; Nakamura, Y.; Yashima, M.; Yoshimura, M.; J. Sol-Gel Sci. Technol.1998, 12, 95.
- 65. Kakihana, M.; Arima, M.; Nakamura, Y.; Yashima, M.; Yoshimura, M.; Chem. Mater. 1999, 11, 438.
- 66. Yamashita, Y.; Yoshida, K.; Kakihana, M.; Uchida, S.; Sato, T.; Chem. Mater. 1999, 11, 61.
- 67. Ribeiro, C.; Mourão, H. A. J. L.; Embrapa Instrumentação Agropecuária, Circular Técnica 39, 2007.
- 68. Malagutti, A. R.; Mourão, H. A. J. L.; Garbin, J. R.; Ribeiro, C.; Appl. Catal., B: Environmental 2009, 90, 205.
- 69. Bernardi, M. I. B.; Soledade, L. E.; Santos, I. A.; Leite, E. R.; Longo, E.; Varela, J. A.; Thin Solid Films 2002, 405, 228.
- 70. Lifshitz, I. M.; Slyozov, V. V.; J. Phys. Chem. Solids 1961, 19, 35.
- 71. Wagner, C. Z.; Zeitschrift Fur Eekektrochemie 1961, 65, 581.
- 72. Oskam, G.; Hu, Z. S.; Penn, R. L.; Pesika, N.; Searson, P. C.; Phys. Rev. E 2002, 66, 011403.
- 73. Penn, R. L.; Banfield, J. F.; Geochim. Cosmochim. Acta 1999, 63, 1549.
- 74. Penn, R. L.; Banfield, J. F.; Science (Washington DC, U. S.) 1998, 281, 969.
- 75. Lee, E. J. H.; Ribeiro, C.; Longo, E.; Leite, E. R.; J. Phys. Chem. B 2005, 109, 20842.
- 76. Ribeiro, C.; Lee, E. J. H.; Longo, E.; Leite, E. R.; ChemPhysChem 2006, 7, 664.
- 77. Ribeiro, C.; Lee, E. J. H.; Longo, E.; Leite, E. R.; ChemPhysChem 2005, 6, 690.
- 78. Lee, E. J. H.; Ribeiro, C.; Longo, E.; Leite, E. R.; Chem. Phys. 2006, 328, 229.
- 79 Ribeiro, C.; Vila, C.; Stroppa, D. B.; Bettini, J.; Mastelaro, V. R.; Longo, E.; Leite, E. R.; J. Phys. Chem. C 2007, 111, 5871.
- 80. Ribeiro, C.; Barrado, C. M.; Camargo, E. R.; Longo, E.; Leite, E. R.; Chemistry (Weinheim) 2009, 15, 2217.
- 81. Zheng, Y.; Erwei, S.; Zhizhan, C.; Wenjun, L.; Xingfang, H.; J. Mater. Chem. 2001, 11, 1457.
- 82. Yang, H. G.; Sun, C. H.; Qiao, S. Z.; Zou, J.; Liu, G.; Smith, S. C.; Cheng, H. M.; Lu, G. Q.; Nature 2008, 453, 638.
- 83. Barnard, A. S.; Curtiss, L. A.; Nano Lett. 2005, 5, 1261.
- 84. Jun, Y-W.; Casula, M. F.; Sim, J-H.; Kim, S. Y.; Cheon, J.; Alivisatos, A. P.; J. Am. Chem. Soc. 2003, 125, 15981.
- 85. Han, S.; Choi, S. H.; Kim, S. S.; Cho, M.; Jang, B.; Kim, D. Y.; Yoon, J.; Hyeon, T.; Small 2005, 1, 812.
- 86. Iijima ,S.; Nature 1991, 354, 56.
- 87. Kasuga, T.; Hiramatsu, M.; Hoson, A.; Sekino, T.; Niihara, K.; Langmuir 1998, 14, 3160.
- 88. Morgan, D. L.; Zhu, H. Y.; Frost R. L.; Waclawik, E. R.; Chem. Mater. 2008, 20, 3800.
- 89. Kasuga, T.; Thin Solid Films 2006, 496, 141.
- 90. Du, G. H.; Chen, Q.; Che, R. C.; Yuan, Z. Y.; Peng, L. M.; Appl. Phys. Lett. 2001, 79, 3702.
- 91. Ma, R.; Bando, Y.; Sasaki, T.; Chem. Phys. Lett. 2003, 380, 577.
- 92. Mao, Y.; Wong, S. S.; J. Am. Chem. Soc. 2006, 128, 8217.
- 93. Jitputti, J.; Suzuki, Y.; Yoshikawa, S.; Catal. Commun. 2008, 9, 1265.
- 94. Marinkovic, B. A.; Jardim, P. M.; Morgado Jr., E.; Abreu, M. A. S.; Moure, G. T.; Rizzo, F.; Mater Res. Bull. 2008, 43, 1562.
- 95. Zhu, K.R.; Yuan, Y.; Zhang, M. S.; Hong, J. M.; Deng, Y.; Yin, Z.; Solid State Commun. 2007, 144, 450.
- 96. Morgado, E.; de Abreu, M. A. S.; Moure, G. T.; Marinkovic, B. A.; Jardim, P. M.; Araújo, A. S.; Mater. Res. Bull. 2007, 42, 1748.
- 97. Ferreira, O. P.; Souza, A. G.; Mendes, J.; Alves, O. L.; J. Braz. Chem. Soc. 2006, 17, 393.
- 98. Geng, J.; Jiang, Z.; Wang, Y.; Yang, D.; Scripta Materialia 2008, 59, 352.
- 99. Geng, J.; Yang, D.; Zhu, J.; Chen, D.; Jiang, Z.; Mater. Res. Bull. 2009, 44, 146.
- 100. Zhu, B.; Li, K.; Zhou, J.; Wang, S.; Zhang, S.; Wu, S.; Huang, W.; Catal. Commun. 2008, 9, 2323.
- 101. Han, C. H.; Hong, D. W.; Kim, I. J.; Gwak, J.; Hana, S. D.; Singh, K. C.; Sens. Actuators, B 2007, 128, 320.
- 102. Niederberger, M.; Garnweitner, G.; Pinna, N.; Neri, G.; Prog. Solid State Chem. 2005, 33, 59.
- 103. Garnweitner, G.; Niederberger, M.; J. Am. Ceram. Soc. 2006, 89, 1801.
- 104. Niederberger, M.; Garnweitner, G.; Chem. Eur. J. 2006, 12, 7283.
- 105. Niederberger, M.; Acc. Chem. Res. 2007, 40, 793.
- 106. Niederberger, M.; Garnweitner, G.; Buha, J.; Polleux, J.; Ba, J. H.; Pinna, N.; J. Sol-Gel Sci. Technol. 2006, 40, 259.
- 107. Ba, J. H.; Fattakhova-Rohlfing, D.; Feldhoff, A.; Brezesinski, T.; Djerdj, I.; Wark, M.; Niederberger, M.; Chem. Mater. 2006, 18, 2848.
- 108. Zhou, S.; Garnweitner, G.; Niederberger, M.; Antonietti, M.; Langmuir 2007, 23, 9178.
- 109. Niederberger, M.; Muhr, H. J.; Krumeich, F.; Bieri, F.; Gunther, D.; Nesper, R.; Chem. Mater. 2000, 12, 1995.
- 110. Pinna, N.; Antonietti, M.; Niederberger, M.; Colloids Surf., A 2004, 250, 211.
- 111. Niederberger, M.; Bartl, M. H.; Stucky, G. D.; Chem. Mater. 2002, 14, 4364.
- 112. Garnweitner, G.; Antonietti, M.; Niederberger, M.; Chem. Commun. 2005, 3, 397.
- 113. Perera S.; Gillan E. G.; Solid State Sci. 2008, 10, 864.
- 114. Li, G.; Gray, K. A.; Chem. Phys. 2007, 339, 173.
- 115. Li, G.; Gray, K. A.; Chem. Mater. 2007, 19, 1143.
- 116. Qin ,W.; Liu, J-J.; Zuo, S-L.; Yu, Y-C.; Hao, Z-P.; J. Inorg. Mater. 2007, 22, 931.
- 117. Xie, R-C.; Shang, J. K.; J. Mater. Sci. 2007, 42, 6583.
- 118. Matijevic, E.; Langmuir 1994, 10, 8.
- 119. Beydoun, D.; Amal, R.; Low, G. K-C.; McEvoy, S.; J. Phys. Chem. B 2000, 104, 4387.
- 120. Watson, S.; Beydoun, D.; Amal, R.; J. Photochem. Photobio., A 2002, 148, 303.
- 121. Lin, Y. S.; Wu, S. H.; Hung, Y.; Chou, Y. H.; Chang, C.; Lin, M. L.; Tsai, C. P.; Mou, C. Y.; Chem. Mater. 2006, 18, 5170.
- 122. Maciel, A. P.; Leite, E. R.; Longo, E.; Varela, J. A.; Cerâmica 2005, 51, 52.
- 123. Libanori, R.; Giraldi, T. R; Longo, E.; Leite, E. R.; Ribeiro, C.; J. Sol-Gel Sci. Technol. 2009, 49, 95.
- 124. Okuya, M.; Nakade, K.; Kaneko, S.; Sol. Energy Mater. Sol. Cells 2002, 70, 425.
- 125. Karunagaran, B.; Kumar, R. T. R.; Kumar, V. S. ; Mangalaraj, D.; Narayandass, S. K.; Rao, G. M.; Mater. Sci. Semicond. Process 2003, 6, 547.
- 126. Vigil, E.; Gonzalez, B.; Zumeta, I.; Docteur, S.; Peiro, A. M.; Gutierrez-Tauste, D.; Dominogo, C.; Domenech, X.; Ayllon, J. A.; J. Cryst. Growth 2004, 262, 366.
- 127. Patil, K. R.; Sathaye, S. D.; Khollam, Y. B.; Deshpande, S. B.; Pawaskwar, N. R.; Mandale, A. B.; Mater. Lett 2003, 57, 1775.
- 128. Biswas, S.; Majumder, A.; Hossain, M. F.; Takahashia, T.; Kubota, Y.; Fujishima, A.; J. Vac. Sci. Technol., A 2008, 26, 678.
- 129. Fang, C.; Chen, Y.; Mater. Chem. Phys 2003, 78, 739.
- 130. Karuppuchamy, S.; Nonomura, K.; Yoshida, T.; Sugiura, T.; Minoura, H.; Solid State Ionics 2002, 151, 19.
- 131. Sankapal, B. R.; Sartale, S. D.; Lux-Steiner, M. C.; Ahmed, E., C. R.; Chimie 2006, 9, 702.
- 132. Giraldi, T. R.; Escote, M. T.; Maciel, A. P.; Longo, E.; Leite, E. R.; Varela, J. A.; Thin Solid Films 2006, 515, 2678.
- 133. Giraldi, T. R.; Escote, M. T.; Bernardi, M. I. B.; Bouquet, V.; Leite, E. R.; Longo, E.; Varela, J. A.; J. Electroceram. 2004, 13, 159.
- 134. Leite, E. R.; Lee, E. J. H.; Ribeiro, C.; Longo, E.; J. Am. Ceram. Soc. 2006, 89, 2016.
- 135. Giraldi, T. R.; Ribeiro, C.; Escote, M. T.; Conti, T. G.; Chiquito, A. J.; Leite, E. R.; Longo, E.; Varela, J. A.; J. Nanosci. Nanotechnol. 2006, 6, 3849.
- 136. Brinker, C. J.; Frye, G. C.; Hurd, A. J.; Ashley, C. S.; Thin Solid Films 1991, 201, 97.
- 137. Zanetti, S. M.; Longo, E.; Varela, J. A.; Leite, E. R.; Mater. Lett. 1997, 31, 173.
- 138. Li, Y.; Dillert, R.; Bahnemann, D.; Thin Solid Films 2008, 516, 4988.
- 139. Chang, C. C.; Chen, J. Y.; Hsu, T. L.; Lin, C. K.; Chan, C. C.; Thin Solid Films 2008, 516, 1743.
- 140. Sayılkan, F.; Asilturk, M.; Tatar, P.; Kiraz, N.; Sener, S.; Arpaç, E.; Sayılkan, H.; Mater. Res. Bull. 2008, 43, 127.
- 141. Negishi, N.; Takeuchi, K.; J. Sol-Gel Sci. Technol. 2001, 22, 23.
- 142. Hsu, Y-Y.; Hsiung, T-L.; Wanga, H. P.; Fukushima, Y.; Wei, Y-L.; Chang, J-E.; Mar. Pollut. Bull. 2008, 57, 873.
- 143. Yuan, Z.; Li, B.; Zhang, J.; Xu, C.; Ke, J.; J. Sol-Gel Sci. Technol. 2006, 39, 249.
- 144. Kitano, M.; Funatsu, K.; Matsuoka, M.; Ueshima, M.; Anpo, M.; J. Phys. Chem. B 2006, 110, 25266.
- 145. Yamaki, T.; Sumita, T.; Yamamoto, S.; Miyashita, A.; J. Cryst. Growth 2002, 237, 574.
- 146. Quan, X.; Ruan, X.; Zhao, H.; Chen, S.; Zhao, Y.; Environ. Pollut. 2007, 147, 409.
- 147. Subramanian, V.; Wolf, E.; Kamat, P. V.; J. Phys. Chem. B 2001, 105, 11439.
- 148. Zheng, J.; Yu, H.; Li, X.; Zhang, S.; Appl. Surf. Sci. 2008, 254, 1630.
- 149. He, C.; Li, X. Z.; Graham, N.; Wang, Y.; Appl. Catal., A 2006, 305, 54.
- 150. Xu, Y.; Shen, M. R.; J. Mater. Process. Technol. 2008, 202, 301.
- 151. Babau, K.; Hatada, R.; Surf. Coat. Technol. 2001, 136, 241.
Datas de Publicação
-
Publicação nesta coleção
26 Nov 2009 -
Data do Fascículo
2009
Histórico
-
Recebido
19 Nov 2008 -
Aceito
29 Abr 2009