Resumo
Amino sugars are chemical compounds that have a sugar backbone, in which one of the hydroxyl groups is replaced by an amino group. Derivatives of amine-containing sugars, such as N-acetylglucosamine, are also considered amino sugars. The synthetic introduction of amino functionalities in a regio- and stereoselective manner ontosugar scaffolds represents a substantial challenge. Most of the modern methods for the preparation of 1-, 2- and 3-amino sugars are those starting from glycals, 2,3-unsaturated O-glycosides epoxy-sugars, keto-sugars among others. This review summarizes recente developments in the current state of knowledge concerning the occurrence, biosynthesis, synthesis and application of amino sugars.
Keywords:
carbohydrate; amino sugars; regioselectivity; diastereoselectivity
Keywords:
carbohydrate; amino sugars; regioselectivity; diastereoselectivity
CONSIDERAÇÕES GERAIS
Os carboidratos e seus derivados são biomoléculas potencialmente úteis nos campos da química e da biologia,11 Teranishi, K.; Carbohydr. Res. 2002, 337, 613. de modo que modificações estruturais nestes compostos podem ocorrer por substituição com nitrogênio, enxofre, fosfato ou cloro, e outros grupos.22 Liu, F. W.; Zhang, Y. B.; Liu, H. M.; Song, X. P.; Carbohydr. Res. 2005, 340, 489. Os aminoaçúcares que são sem dúvida os mais importante açúcares modificados existentes, são os constituintes fundamentais33 Taylor, C. M.; Tetrahedron 1998, 54, 11317.,44 Tu, D.; Blaha, G.; Moore, P. B.;. Steitz, T. A.; Cell 2005, 121, 257. de muitos compostos biologicamente ativos, como antibióticos55 Mankin,A. S.; Curr. Opin. Microbiol. 2008, 11, 414.,66 Zhang, Z.; Fukuzaki, T.; Myer, A. G.; Angew. Chem., Int. Ed. 2016, 55, 523. e biopolímeros.77 Zhu, R.; Lin, Y.-S.; Lipp, J. S.; Meador, T. B.; Hinrichs, K.-U.; Biogeosciences 2014, 11, 4869. Aminoaçúcares são definidos como aldoses ou cetoses que têm seu grupo hidroxila substituído por grupo amino nos C2, C3 ou C4 do anel hexose ou pentose, exceto no carbono anomérico.88 Skarbek, K.; Milewska, M. J.; Carbohydr. Res. 2016, 434, 44.,99 Fischer, E.; Leuchs, H.; J. Chem Pub Soc. Eur. 1902, 35, 3787. As propriedades físico-químicas dessas moléculas podem afetar fortemente suas atividades biológicas. Esses compostos são cruciais para o bem estar da maioria dos organismos, incluindo humanos, porque desempenham papéis essenciais na estrutura e função de oligossacarídeos, polissacarídeos e glicoproteínas biologicamente importantes.1010 El Ashry, E. S. H.; Aly, M. R. E. P.; Pure Appl. Chem. 2007, 79, 2229.,1111 Arsequell, G.; Valencia, G.; Tetrahedron: Asymmetry 1999, 10, 3045. A presença de um amino grupo em um resíduo de monossacarídeo permite a protonação e geração de um íon amônio; o grupo acetamido, embora muito menos básico que uma amina, transmite muita polaridade à molécula em questão. Sabe-se, por exemplo, que a estereoquímica relativa dos grupos funcionais em aminoaçúcar natural e não natural desempenha um papel importante no perfil de atividade das antraciclinas.1212 De Freitas Filho, J. R.; Srivastava, R. M.; Carbohydr. Res. 2003, 338, 673.
Os aminoaçúcares podem ser obtidos através de síntese total ou por introdução nos açúcares de um grupo funcional amino. Alguns deles foram abordados na literatura no período compreendido entre 1963 e 2018.1313 Salton, M. R. J.; Annu. Rev. Biochem. 1965, 34, 143.
14 Ashwell, G.; Annu. Rev. Biochem. 1964, 33, 101.
15 Dutcher, J. D.; Adv. Carbohydr. Chem.
1963, 18, 259.
16 Pfrengle, F.; Reissig, H-U.; Chem. Soc. Rev. 2010, 39, 549.
17 Kirschning, A.; Jesberger, M.; Schoning, K.; Synthesis
2001, 4, 507.
18 Xie, J; Carbohydrate Res.
2003, 338, 399.
19 Levites-Agababa, E.; Menhaji, E.; Perlson, L. N.; Rojas, C. M.; Org. Lett.
2002, 4, 863.-2020 Chen, N.; Xie, J.; Molecules
2018, 23, 641. Uma revisão mais recente, descreve a ocorrência, biossíntese e métodos de síntese de aminoaçúcares.88 Skarbek, K.; Milewska, M. J.; Carbohydr. Res. 2016, 434, 44. Em 2018, Chen e Xie2020 Chen, N.; Xie, J.; Molecules
2018, 23, 641. relataram em um artigo de revisão a síntese e aplicações biológicas de O-amino acúcares e nucleosídeos. Outras publicações voltadas para síntese e aplicações biológicas de aminoaçúcares de aminoaçúcares e seus derivados, são descritos na literatura.1616 Pfrengle, F.; Reissig, H-U.; Chem. Soc. Rev. 2010, 39, 549.,2121 Davidson, M. H.; McDonald, F. E.; Org. Lett.
2004, 6, 1601.
22 Velvadapu, V.; Andrade, R. B.; Carbohydr. Res. 2008, 343, 145.
23 Paixão, L.; Caldas, J.; Kloosterman, T. G.; Kuipers, O. P.; Vinga, S.; Neves, A. R.; Front Microbiol. 2015, 6, 1041.-2424 Kent, P. W.; Whitehouse, M. W.; Biochemistry of the aminosugars, Academic Press: New York, 1955. Os membros mais comuns dessa classe de compostos são 2-amino-2-desoxi-D-glicopiranose (D-glicosamina ou quitosamina 1), 2-acetamido-2-desoxi-D-glicopiranose (2) e 2-amino-2-desoxi-D-galactopiranose (3) (Figura 1).
Portanto, neste trabalho, resumimos o conhecimento atual sobre a química, ocorrência natural, biossíntese, síntese e aplicações de aminoaçúcares.
OCORRÊNCIA NATURAL DE AMINOAÇÚCARES
Aminoaçúcares são membros de uma classe de compostos de grande diversidade e abundância na natureza. O termo “açúcar aminado” refere-se a um derivado de hidrato de carbono em que um ou mais dos grupos hidroxila foram substituídos por um grupo amino. O grupo amino pode ser livre ou substituído. Nesta Seção, é apresentada uma visão geral da ocorrência natural de aminoaçúcares.
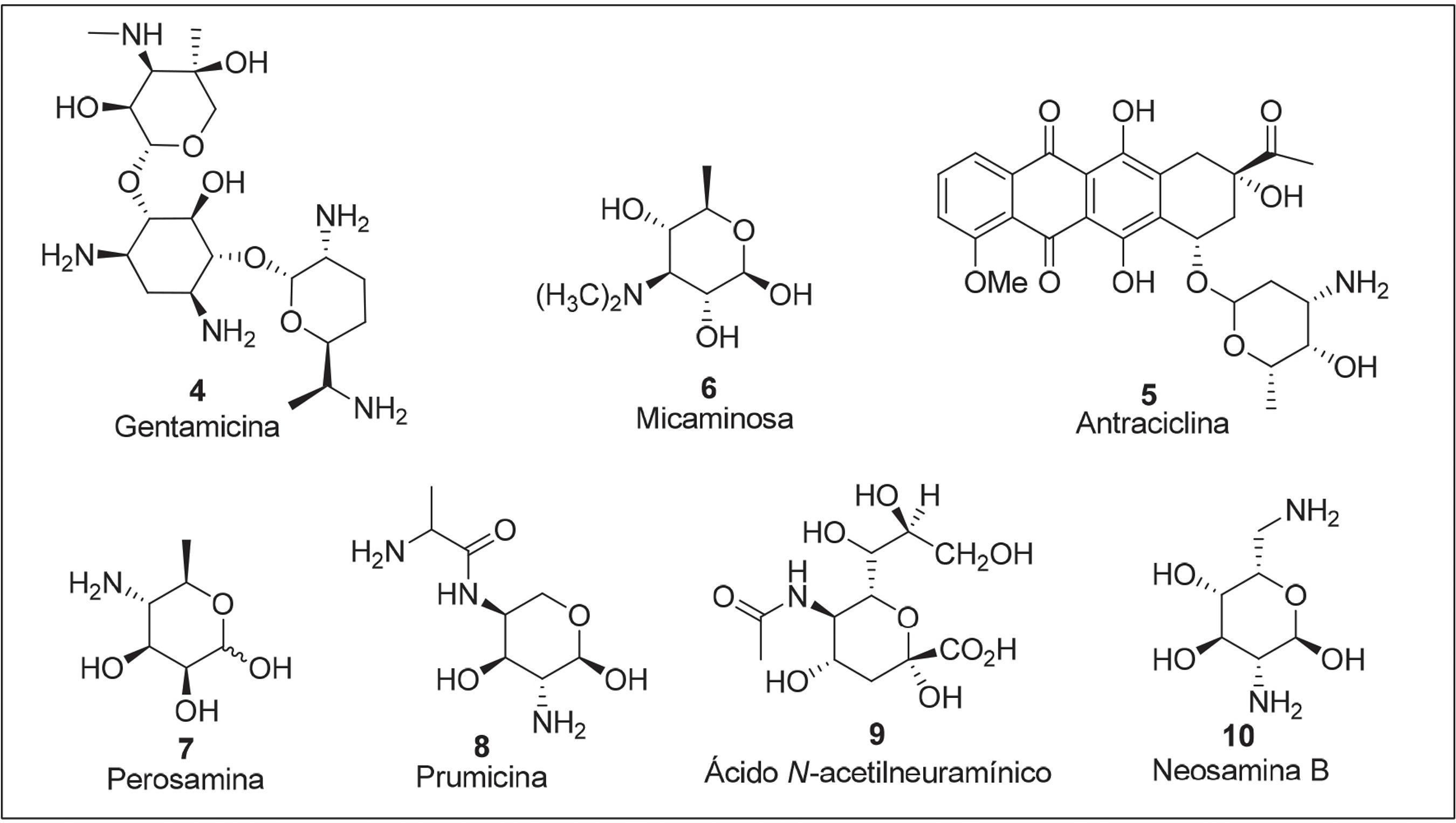
Por exemplo, a 2-amino-2-desoxi-D-glicose (D-glicosamina ou quitosamina 1)2424 Kent, P. W.; Whitehouse, M. W.; Biochemistry of the aminosugars, Academic Press: New York, 1955. é muito abundante na natureza, particularmente presente no polissacarídeo quitina, em que pode ser encontrada na forma de derivados N-Acetilados 2.2525 Ledderhose, G.; Zeitschr, F.; Physiol. Chem. 1878, 2, 213.
26 Schmiedeberg, O.; Arch. F. Exp. Path. U. Pharm. 1891, 28, 355.-2727 Moiler, F.; Z. Biol. 1901, 42, 468. O composto 2-amino-2-desoxi-D-galactose 3 (D-galactosamina)2828 Levene, P. A.; LaForge, F. B.; J. Biol. Chem. 1914, 18, 123. é bastante comum e é um monossacarídeo constituinte da dermatana e do sulfato de condroitina, polissacarídeos encontrados nos tecidos e cartilagem de mamíferos. Essas substâncias são nomeadas segundo o açúcar do qual são derivadas pelo uso do prefixo “aminodesoxi”. Além de grupo amino na posição 2 do carboidrato, encontra-se também a ocorrência de grupos amino nas posições 3, 4, 5 ou 6 (Figura 2). Açúcares do tipo 3-amino são encontrados frequentemente na natureza como constituintes de gentamicina 42929 Gutiérrez-Moreno, N. J.; Medranob, F.; Yatsimirsky, A. K.; Org. Biomol. Chem. 2012, 10, 6960. e do antibiótico antraciclina 5.3030 McGowan, J. V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J. M.; Yellon, D. M.; Cardiovasc. Drugs Ther. 2017, 1, 63. A micaminose, constituinte de antibióticos como a leucomicina, magnamicina, e outros membros do grupo espiromicina,3131 Barata, L. E. S.; Cienc. Cult. (Sao Paulo) Supl.
1980, 32, 449. é identificada como 3,6-didesoxi-3-dimetilamino-D-glicose 6. Os açúcares 4-amino-4,6-didesoxihexoses e seus derivados são constituintes de um grande número de antibióticos.3232 Lee, C.-H.; Schaffner, C.; Tetrahedron Lett. 1966, 47, 5837. O componente aminoaçúcar (perosamina) do antibiótico heptaênico perimicina é reconhecido através de evidência química como 4-amino-4,6-didesoxi-D-manose 7.3232 Lee, C.-H.; Schaffner, C.; Tetrahedron Lett. 1966, 47, 5837.,3333 Els, M. J.; Ganem, B.; Carbohydr. Res. 1988, 176, 316. O composto prumicina, nomeado como 4-(D-alanilamino)-2-amino-2,4-didesoxi-L-arabinose 8, além da atividade antibiótica, tem atividade antimoral3434 Okubo, S.; Nakamura, N.; Morinoto, M.; Mineura, K.; Marumo, H.; Omura, S.; The Journal of Antibiotics
1980, 33, 221. e foi sintetizado a partir de glicina e L-serina.3535 Hamada, Y.; Shioiri, T.; Tetrahedron Lett.
1982, 23, 1193.
Os ácidos nonulosamínicos (ácidos neuramínicos), por exemplo, são derivados de 5-amino-5-desoxinonose encontrado usualmente na forma combinada no mucolipídeos ou mucopolissacarídeos em animais. Um exemplo é o ácido 5-acetamido-3-5-didesoxi-D-glicero-D-galacto-2-nonulosônico (ácido N-acetilneuramínico) 9 (Figura 2), conhecido como ácido siálico e encontrado em polissacarídeos de muitos animais e bactérias.3636 Troy, F. A.; Encyclopedia Biol. Chem.
2004, 3, 407.
37 Kleene, R.; Schachner, M.; Nat. Rev. Neurosci. 2004, 5, 195.
38 Dumitriu. S.; Polysaccharides: Structural Diversity and Functional,Versality: 2nd ed., Marcel Dekker: New York, 2005, Cap. 30.
39 Bonfanti, L.; Prog. .Neurobiol.
2006, 80, 129.
40 Gascon, E.; Vutskits, L.; Kiss, J. K.; Brain Res. Rev. 2007, 56, 101.
41 Miyata, S.; Sato, C.; Kitajima, K.; Trends Glycosci. Glyc.
2007, 19, 85.-4242 Janas, T.; Janas, T.; Biochim. Biophys. Acta
2011, 1808, 2923.
O ácido 5-acetamido-3,5-didesoxi-D-glicero-D-galacto-2-nonulosônico foi sintetizado com 51% de rendimento a partir do ácido nonônico. Na literatura, há uma descrição detalhada sobre o espectro de RMN 1H e de 13C do Neu5Ac em solução de óxido de deutério.4343 Kimio, F.; Trends Glycosci. Glycotechnol. 2004, 89, 143. A conformação do anel piranosídico de Neu5Ac, é do tipo 1C4, e a configuração β é estabilizada no grupo hidroxila da posição anomérica. Para caracterização da estrutura do composto 9, foi realizada análise elementar, rotação específica, espectro de infravermelho e cristalografia de raios-X.4343 Kimio, F.; Trends Glycosci. Glycotechnol. 2004, 89, 143. Outros aminoaçúcares derivados têm mostrado interessantes propriedades farmacológicas, como por exemplo o composto 2,6-diamino-2,6-didesoxi-L-idose 10 (neosamina B) que é um constituinte do antibiótico paromomicina e neomicina B4444 Huang, F.; Spiteller, D.; Koorbanally, N. A.; Li, Y.; Llewellyn, N. M.; Spencer, J. B.; ChemBioChem 2007, 8, 283. é um exemplo de aminoaçúcar com grupo amino na posição 6 (Figura 2).
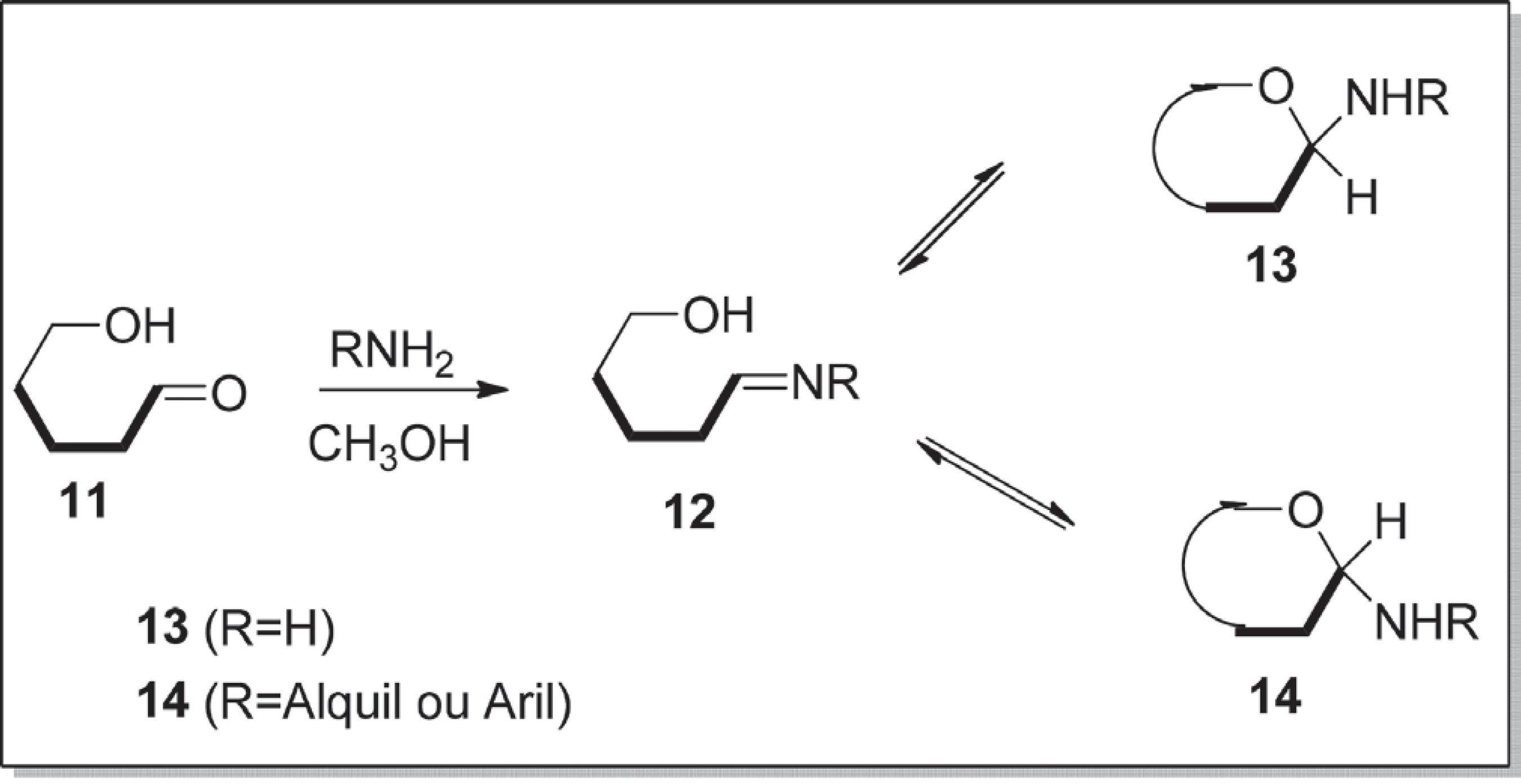
Carboidratos nos quais um grupo amino substitui o –OH anomérico são chamados de glicosilaminas ou N-glicosídeos e podem ocorrer na forma de um anel de cinco ou seis membros e na configuração α- e β-anomérica (compostos 13 e 14). As aldoses sofrem reações de condensação com amônia, aminas primárias ou secundárias para fornecer glicosilaminas. Um exemplo desse tipo de reação é a condensação da aldose 11 com amônia e aminas primária (Esquema 1),4545 Ellis, G. P.; Honeyman, J.; J. Chem. Soc. 1952, 1490. produz um aduto de imina instável 12, também chamado de base de Schiff, o qual é ciclizada para originaros α e β-anômeros 13 e 14. Uma análise mais recente dos mecanismos de reação da formação de glicosilamina identificou várias rotas para a formação de produtos.4646 Hermanson, G. T.; Bioconjugate Techniques 3rd ed., Academic Press: Cambridge 2013, cap. 2.
Outros aminoaçúcares de origem natural são frequentemente constituintes de diferentes produtos do metabolismo secundário, incluindo antibióticos. A micosamina está presente nos antibióticos antifúngicos, anfotericina B e nistatina.4747 Anderson, J. W.; Nicolosi, R. J.; Borzelleca, J. F.; Food Chem. Toxicol. 2005, 43, 187.,4848 Brautaset, T.; Sletta, H.; Nedal, A.; Borgos, S. E.; Degnes, K. F.; Bakke, I.; Volokhan, O.; Sekurova, O. N.; Treshalin, I. D.; Mirchink, E. P.; Dikiy, A.; Ellingsen, T. E.; Zotchev, S. B.; Chem Biol. 2008, 15, 1198. A ribosamina é um componente da puromicina, a desosamina está presente na eritromicina, a daunosamina ocorre no anticâncer antraciclina daunomicina e diferentes açúcares aminados e seus derivados são encontrados nos antibióticos antibacterianos aminoglicosídeos. Em cada caso, a porção aminoaçúcar é importante para a atividade biológica desses antimicrobianos.4949 Muhizi, T.; Grelier, S.; Coma, V.; J. Agric. Food Chem. 2009, 57, 8770.
ROTAS BIOSSINTÉTICAS DE AMINOACÚCARES
Os aminoaçúcares podem ser obtidos através de rotas sintéticas e biossintéticas. Na rota biossintética, a conversão de um monossacarídeo – geralmente na forma de açucar fosfatado ou de nucleotídeo glicosilado – é catalisada por uma enzima aminotransferase PLP-dependente ou amidotransferase PLP-independente, de maneira estereoespecífica. Por outro lado, a sua síntese será possível através de precursores, tais como álcoois, aldeídos, ácidos carboxílicos, ésteres e lactonas ou aminoácidos ou pela introdução regio- e estereoespecífica de uma funcionalidade amino em açúcares apropriados, ou substrato derivado de açúcar por substituição nucleofílica ou adição. Inicialmente discutiremos as rotas biossintéticas para obtenção de aminoaçúcares derivados do metabolismo primário e secundário numa abordagens diferentes e mais completa do que foi descrito por Skabec e Milewska.88 Skarbek, K.; Milewska, M. J.; Carbohydr. Res. 2016, 434, 44.
Aminoaçúcares derivados do metabolismo primário
Uma rota biossintética para obtenção de aminoaçúcares no metabolismo primário envolve a transferência do grupo amino de um doador de aminoácido (geralmente L-glutamato ou L-glutamina) para a função ceto de um derivado de cetose na forma aberta ou para o átomo de carbono ceto gerado na reação anterior após oxidação de um C-OH da forma cíclica de um derivado da aldose. A transferência de grupo amino é catalisada pela enzima aminotransferase PLP-dependente ou amidotransferase PLP-independente, de maneira estereoespecífica. O substrato aceitador do grupo amino é geralmente um açúcar fosfatado ou um nucleotídeo glicosilado. A única reação desse tipo no metabolismo primário dá origem a α-D-glicosamina-6P (21) a partir de β-D-frutose-6P (15). Essa reação é catalisada por L-glutamina: β-D-frutose-6P amidotransferase (isomerização da hexose), cujo nome trivial é glicosamina-6P (GlcN-6-P) sintase. A enzima é amplamente distribuída na natureza e está presente em quase todos os organismos vivos. Segundo Adam,5050 Adam, R. D.; Clin. Microbiol. 2001, 14, 447. no protozoário Giardia lamblia, o GlcN-6P é formado a partir de Fru-6-P e amônia sobre catálise com GlcN-6P desaminase, uma enzima catabólica em todos os outros organismos.
A GlcN-6-P sintase não requer nenhuma coenzima e catalisa reação complexa, irreversível, que envolve transferência do grupo amida de L-glutamina para β-D-Fru-6P e subsequente isomerização da cetose para o intermediário frutoseimina 19, Esquema 2.
De acordo com Milewski et al.,5151 Milewski, S.; Gabriel, I.; Olchowy, J.; Yeast 2006, 23, 1. a GlcN-6-P formada na reação catalisada pela GlcN-6-P sintase é posteriormente convertida em UDP-GlcNAc, em uma série de três reações consecutivas, conhecida como a via Leloir. Por outro lado, a UDP-GlcNAc dá origem aos respectivos nucleotideos glicosilados, tais como ManNAc e GalNAc, formados na reação catalisada pela uridina difosfato-N-acetilglicosamina-2-epimerase e uridina difosfato-N-acetilglicosamina-4-epimerase, respectivamente.5252 Kikuchi, K.; Tsuiki, S.; Biochim. Biophys. Acta. 1973, 327, 193.,5353 Chen, X.; Varki, A.; ACS Chem. Biol. 2010, 5, 163. O ManNAc é um precursor do ácido siálico (Neu5Ac), formado após condensação catalisada por aldolase de ManNAc e piruvato (Esquema 3).
Rota biossintética de Neu5Ac após condensação catalisada por aldolase de ManNAc e piruvato
Uma abordagem mais comum para a síntese enzimática de Neu5Ac envolve a preparação química da N-acetilmanosamina modificada (ManNAc), derivados 23, e a sua conversão no correspondente derivado Neu5Ac utilizando a enzima Neu5Ac aldolase conforme representado no Esquema 4. Convém destacar que a enzima Neu5Ac aldolase catalisa a clivagem reversível do Neu5Ac em piruvato e ManNAc.5454 Adak, A. K.; Yu, C. C.; Liang, C. F.; Lin, C. C.; Curr. Opin. Chem. Biol. 2013, 17, 1030.
Por outro lado, a formação da ligação carbono-carbono entre o ManNAc e o piruvato depende não apenas da proximidade e orientação dos dois átomos de carbono ligados, mas também da transferência de um hidrogênio para o oxigênio da carbonila da aldose do ManNAc.
Aminoaçúcares derivados do metabolismo secundário
A biossíntese de aminoaçúcares no metabolismo secundários é realizada por rotas metabólicas específicas com estreita relação com as rotas biossintéticas no metabolismo primário. Essas rotas são interconectadas, ou seja, as rotas que sintetizam metabólitos primários fornecem moléculas que são utilizadas como precursoras nas principais rotas de síntese de metabólitos secundários.
Ao contrário da biossíntese de aminoacúcares no metabolismo primário, que deriva da aminação catalisada pela D-glicosamina-6P sintase de D-frutose-6P, a biossíntese de aminoacúcares no metabolismo secundário envolve a introdução da funcionalidade amino por uma enzima aminotransferase PLP-dependente. Essa enzima participa da biossíntese de 3-amino-3-desoxi-D-glicose (kanosamina).5555 Vetter, N. D.; Langill, D. M.; Anjum, S.; Boisvert-Martel, J.; Jagdhane, R. C.; Omene, E.; Zheng, H.; van Straaten, K. E.; Asiamah, I.; Krol, E. S.; Sanders, D. A.; Palmer, D. R.; J. Am. Chem. Soc. 2013, 135, 5970. De acordo com o Esquema 4, a glicose-6P 24 é convertida em três etapas para kanosamina pela ação da enzima NtdC, uma 3-desidrogenase de glicose-6P, uma enzima NtdA, um 3-oxo-glicose-6-P dependente de fosfato de piridoxal (PLP) e uma enzima denominada de NtdB, uma fosfatase kanosamina-6P fosfatase. A aminação de 3-oxo-D-glicose-6-fosfato nessa via é catalisada pela NtdA aminotransferase. Essa enzima foi isolada e sua estrutura foi determinada.5656 van Straaten, K. E.; Ko, J. B.; Jagdhane, R.; Anjum, S.; Palmer, D. R.; Sanders, D. A.; J. Biol. Chem. 2013, 288, 34121.
A kanosamina é produzida por Bacillus subtilis, Bacillus circulans e Bacillus pumilus exibe efeito inibidor do crescimento contra Staphylococcus aureus e Klebsiella pneumoniae.5757 Iwai, Y.; Tanaka, H.; Oiwa, R.; Shimizu, S.; Omura, S.; Biochim. Biophys. Acta 1977, 498, 223.,5858 Janiak, A.; Milewski, S.; Med. Mycol. 2001, 39,401. O mecanismo de ação antimicrobiana envolve o transporte pela hexose permease, fosforilação a kanosamina-6-fosfato (K6P) e inibição GlcN-6-P sintase pela K6P.5858 Janiak, A.; Milewski, S.; Med. Mycol. 2001, 39,401. Em 2011, Floss e colaboradores5959 Floss, H. G.; Yu, T. W.; Arakawa, K.; J. Antibiot. 2011, 64, 34. descreveram que a kanosamina e a UDP-kanosamina são também os intermediários específicos na biossíntese do 3-amino-5-hidroxibenzoato, um precursor dos antibióticos mitomicina e ansamicina, incluindo a rifamicina B. Além de ser o precursor biossintético da via biossintética 3-amino-5-hidroxibenzoato, a kanosamina também é a fonte de nitrogênio para a via amino shiquimato.6060 Arakawa, K.; Müller, R.; Mahmud, T.; Yu, T.W.; Floss, G.; J. Am. Chem. Soc. 2002, 124, 10644.
A melhor rota biossintética para a produção de kanosamina envolve a fosforilação da glicose para a glicose 1-fosfato, seguida pela pirofosforilação em UDP-glicose. A UDP-glicose é então oxidada em UDP-3-oxo-D-glicose. A transaminação de UDP-3-oxo-D-glicose produz UDP-kanosamina, que é então hidrolisada para formar kanosamina.
A biossíntese de kanosamina em Streptomyces envolve a fosforilação de glicose em glicose-1-fosfato, seguida de pirofosforilação em UDP-glicose. O nucleotídeo glicosilado é posteriormente oxidado a UDP-3-oxo-D-glicose.6161 Guo, J. T.; Frost, J. W.; J. Am. Chem. Soc. 2002, 124, 10642. A transaminação de UDP-3-oxo-D-glicose produz UDP-kanosamina, que é finalmente hidrolisada para produzir kanosamina.6262 Park, J. W.; Park, S. R.; Nepal, K. K.; Han, A. R.; Ban, Y. H.;Yoo, Y. J.; Kim, E. M.; Kim, D.; Sohng, J. K.; Yoon, Y. J.; Nat. Chem. Biol. 2011, 7, 843.
Outros aminoacúcares provenientes do metabolismo secundário microbiano são componentes de muitos antibióticos. Por exemplo, a D-desosamina, também conhecida como 3-(dimetilamino)-3,4,6-trideoxi-D-glicosee presente no antibiótico eritromicina é formada a partir da glicose em uma via biossintética de seis etapas envolvendo aminação de TDP-3-oxo-6-desoxi-D-glicose por DesV aminotransferase6363 Burgie, E. S.; Thoden, J. B.; Holden, H. M.; Protein Sci. 2007, 16, 887. seguido de dimetilação catalisada por N,N-dimetiltransferase DesVI (Esquema 5).6464 Burgie, E. S.; Holden, H. M.; Biochemistry 2008, 47, 3982. A produção de dTDP-desosamina inicia com a ligação de α-D-glicose-1-fosfato ao dTMP catalisada por DesIII. O passo seguinte na remoção do grupo hidroxila do C6 do açúcar é a oxidação do grupo hidroxila do C4 para uma funcionalidade ceto através da ação da enzima DesIV. Em seguida, a enzima DesI promove a aminação no C4 do açúcar. Para que essa etapa aconteça é necessário primeiramente a conversão de PLP em PMP usando glutamato como fonte de nitrogênio, seguida pela aminação do C4 do acúcar, com o grupo amino na posição equatorial. Após a aminação por DesI, a enzima seguinte na via, DesII, remove o grupo amino da posiçao C-4 e oxida o grupo hidroxila de C-3 à cetona. Ainda de acordo com o Esquema 5, existe uma segunda enzima PLP-dependente na via, a DesV, que adiciona um grupo amino no C-3 e na posição equatorial.
Via biossintética de seis etapas envolvendo aminação de TDP-3-oxo-6-desoxi-D-glicose por DesV aminotransferase
A estrutura de DesI foi comprovada através de difração de raios-X, cuja resolução foi realizada com o complexo dTDP-4-amino-4,6-didesoxiglicose.
AD-perosamina (4-amino-4,6-didesoxi-D-manose), aminoacúcar derivado do metabolismo secundário e presente em Vibrio cholerae, é biossintetizada como um derivado ligado ao GDP da GDP-4-ceto-6-desoxi-D-manose, em uma reação catalisada pela enzima perosamina-sintase.6565 Cook, P. D.; Holden, H. M.; Biochemistry 2008, 47, 2833. Por outro lado, a aminação da GDP-3-ceto-6-desoxi-D-manose pela GDP-3-ceto-6-desoxi-D-manose 3-aminotransferase NysDII origina a GDP-micosamina (GDP-3-amino-3,6-dideoxi-D-manose), o qual é um precursor da porção D-micosamina, presente num antibiótico antifúngico denominado nistatina produzido por Streptomyces noursei.6666 Nedal, A.; Sletta, H.; Brautaset, T.; Borgos, S. E. F.; Sekurova, O. N.; Ellingsen, T. E.; Zotchev, S. B.; Appl. Environ. Microbiol. 2007, 73, 7400. Segundo Otten e colaboradores,6767 Otten, S. L.; Gallo, M. A.; Madduri, K.; Liu, X.; Hutchinson, C. R.; J. Bacteriol. 1997, 179, 4446. a TDP-daunosamina, um precursor da porção daunosamina no antibiótico anti-daunorrubicina, é derivada da 3,4-diceto-2-desoxi-D-ramnose, que é aminada em C3.
A estrutura e configuração absoluta da L-daunosamina foi determinada principalmente por sua similaridade espectral com a rodosamina e 2-desoxi-L-fucose, ambas com configuração de L-lyxo. Um passo fundamental na elucidação da estrutura foi a degradação oxidativa do derivado N-benzoila da L-daunosamina a partir do ácido L-aspártico.
Outros 3-amino-2,3,6-trideoxi-hexoses presentes em antraciclinas ou vancomicinas, incluindo L-ristosamina, L-acosamina e L-vancosamina, são formadas a partir das respectivas 3-ceto-2,6-didesoxi-hexoses de maneira similar.6868 Hauser, F. M.; Ellenberger, S. R.; Chem. Rev. 1986, 86, 35. Por outro lado, Guo et al.6969 Guo, Z.; Li, J.; Qin, H.; Wang, M.; Lu, X.; Li, X.; Chen, Y.; Angew. Chem., Int. Ed. Engl. 2015, 54, 5175. relatam que a D-gulosamina (2-amino-2-desoxi-D-gulose) presente em aminoglicosídeos atípicos, é proveniente da epimerização e desacetilação da N-acetil-D-galactosamina. O UDP-N-metil-D-glicosamina-6-fosfato é identificado como um precursor para a biossíntese de N-metil-L-glicosamina, um componente da estreptomicina.7070 Kumagai, A. H.; Yagita, A.; Akamatsu, N.; J. Antibiot. 1982, 34, 1571. A Figura 3, a seguir, sumariza exemplos de alguns aminoaçúcares derivados do metabolismo secundário.
SÍNTESE DE AMINOAÇÚCARES
Alguns métodos relatados para síntese de aminoaçúcares envolvem as ligações dupla dos glicais 48, obtidos a partir do composto 47, e dos hexenopiranosídeos 49 (Esquema 6), que representam excelentes materiais de partida devido ao seu baixo custo e disponibilidade comercial.7171 Mirabella, S.; Cardona, F.; Goti, A.; Org. Biomol. Chem. 2016,
14, 5186. Sua obtenção se dá após abertura de derivados epoxi-açúcares por aminas,88 Skarbek, K.; Milewska, M. J.; Carbohydr. Res. 2016, 434, 44. redução de derivados de oxima e azida,7272 Dahl, R. S.; Finney, N. S.; J. Am. Chem. Soc.
2004, 126, 8356. reação de adição a ligações duplas de glicais7373 Buttar, S.; Caine, J.; Goné, E.; Harris, R.; Gillman , J.; Atienza, R.; Gupta , R. K. M.; Jain, L.; Abascal, N. C.; Levine, Y.; Repka, L. M.; Rojas, C. M.; J. Org. Chem., 2018, 83, 8054.
74 Ansari, A. A.; Reddy, Y. S.; Vankar, Y. D.; Beilstein J. Org. Chem. 2014, 10, 300.-7575 Seeberger, P. H.; Roehrig, S.; Schell, P.; Wang, Y.; Christ, W. J.; Carbohydr. Res. 2000, 328, 61. e de glicosídeos 2,3-insaturados7676 Liu, J.; Di Bussolo, V.; Gin, D. Y.; Tetrahedron Lett.
2003, 44, 4015. e cicloadição à cetoaçúcares, dentre outros.7777 Schmidt, R. R.; Vankar, Y. D.; Acc. Chem. Res.
2008, 41, 1059.
Síntese de aminoaçúcares a partir de epóxi-açúcares
Em 1993, Roger e colaboradores7878 Rissé, S.; Roger, P.; Monneret, C.; J. Carbohydr. Chem. 1993, 12, 1105. descreveram a síntese do 3-Amino-3,4-dideoxi-β-D-xilo-hexapiranosideode metila 59 com azida de sódio (Esquema 7). Nessa reação houve a formação de dois intermediários 57 e 58, os quais foram obtidos na proporção de 3:2. Esta mesma reação de síntese a partir do epóxido 56 também foi mencionado por Ferrier e Collins.7979 Ferrier, R. J.; Collins, P. M.; Monosaccharides: Their chemistry and their roles in natural products. Wiley & Sons: Chichester, 1995.
Síntese do 3-Amino-3,4-dideoxi-β-D-xilo-hexapiranosideo de metila usando com azida de sódio
Okazaki e colaboradores8080 Okazaki, H.; Hanaya, K.; Shoji, M.; Hada, N.; Sugai, T.; Tetrahedron 2013, 69, 7931. propuseram a síntese de dois intermediários contendo anéis de três membros, o epóxido 60, e em seguida o isômero syn-60 foi subsequentemente aberto usando azida de sódio (Esquema 8) para dar a mistura dos regioisômeros 61ab (74:28).
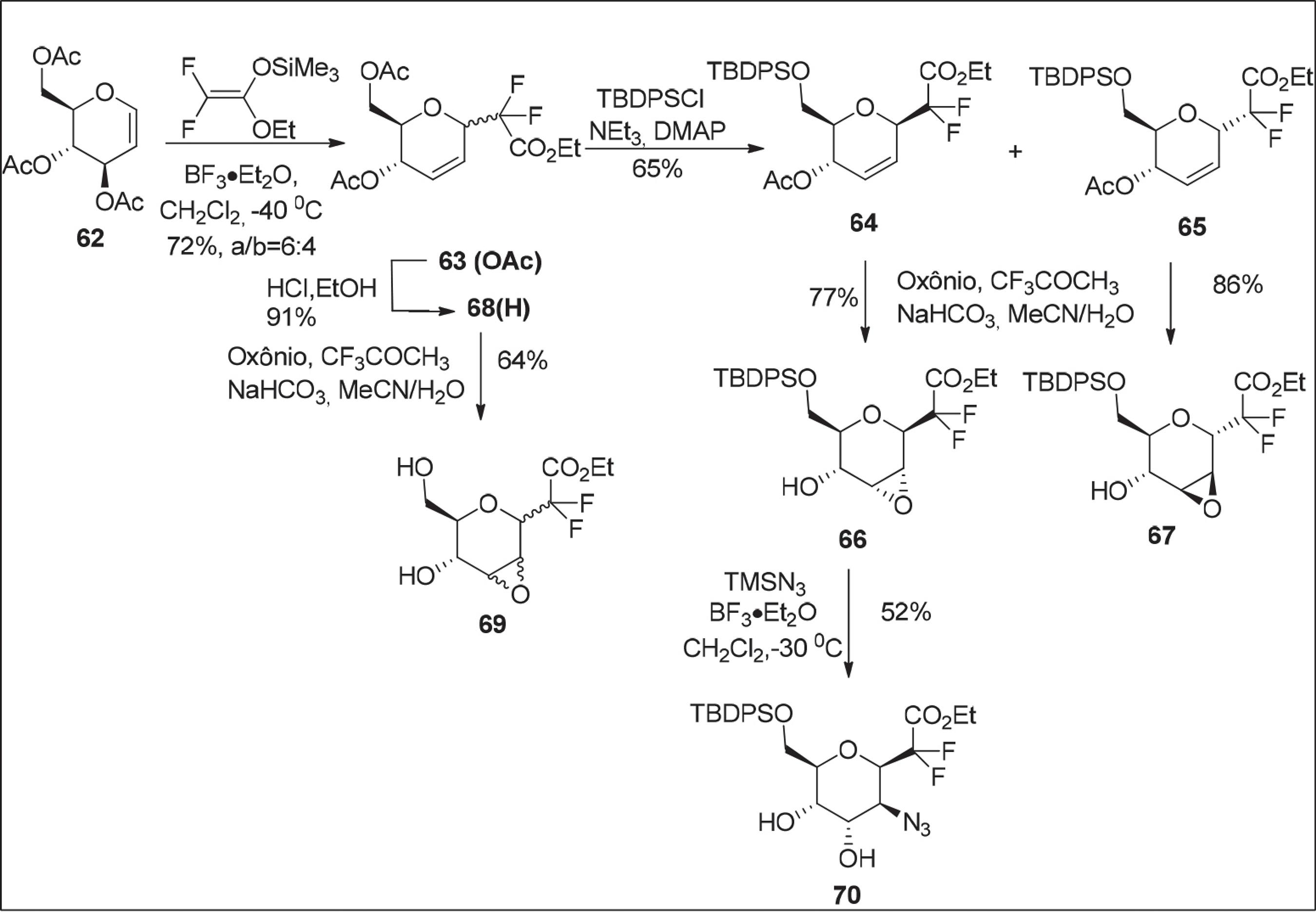
Em 2009, Poulain e colaboradores8181 Poulain, F.; Serre, A.-L.; Lalot, J.; Leclerc, E.; Quirion, J.-C.; J. Org. Chem. 2008, 73, 2435. relataram a reação de um difluoroceteno acetal sililado ao D-glical 62 para produzir o C-glicosídeo 2,3-insaturado 63, o qual foi submetido a uma reação de desproteção para fornecer 68 e uma reação de proteção para fornecer os α,β-anômeros 64 e 65. Segundo os autores, a partir das reações de desproteção/proteção, duas sequências de reações foram planejadas para obtenção de aminoacúcares: a) reação epoxidação da ligação dupla de 64, 65 e 68 e b) reação de abertura de anel dos epóxidos com nucleófilos de nitrogênio (TMSN3) para fornecer o composto 70 em rendimento de 52% (Esquema 9).8282 Poulain, F.; Leclerc, E.; Quirion, J.-C.; Tetrahedron Lett. 2009, 50, 1803.
Ainda conforme os autores, a oxidação de 64 e 65 com o (trifluorometil) dioxirano, gerado in situ a partir de trifluoroacetona e oxônio produziu os epóxidos 66 e 67 com completa diastereosseletividade, ou seja, com o átomo de oxigênio entrando trans em ralação ao substituinte anomérico. Destes, apenas o diastereoisômero 66 foi submetido à abertura de anel usando com trimetilsililazida em presença de um ácido de Lewis, para fornecer o 2-desoxi-2-azido-C-glicosídeo 70 como um único diastereoisômero. Outras rotas sintética para produzir aminoaçúcares via epoxi-açúcares são descritas na literatura.8383 Okazaki, H.; Hanaya, K.; Shoji, M.; Hada, N.; Sugai, T.; Tetrahedron
2013, 69, 7931.
84 Sobti, A. Sulikovski, G. A.; Tetrahedron Lett. 1994, 35, 8259.
85 Castilla, J.; Risquez, R.; Cruz, D.; Higaki, K.; Nanba, E.; Ohno, K.; Suzuki, Y.; Diaz, Y.; Mellet, C. O.; Garcia Fernandez, J. M.; Castillon, S.; J. Med. Chem. 2012, 55, 6857.-8686 Ji, L.; Zhang, D.; Zhao, Q.; Hu, S.; Qian, C.; Chen, X.-Z.; Tetrahedron
2013, 69, 7031.
Síntese de aminoaçúcares a partir de glicais e/ou de glicosídeos 2,3-insaturados
Os produtos de partidas utilizados nestas reações de síntese são 1,2-glicais ou glicosídeos 2,3-insaturados protegidos,8787 Parker, K. A.; Chang, W.; Org. Lett.
2005, 7, 1785.
88 Parker, K. A.; Chang, W.; Org. Lett.
2003, 5, 3891.
89 Alcázar, E.; Pletcher, J. M.; McDonald, F. E.; Org. Lett.
2004, 6, 3877.
90 Koo, B. S.; McDonald, F. E.; Org. Lett.
2007, 9, 1737.-9191 Davidson, M. H; McDonald, F. E.; Org. Lett.
2004, 6, 1601. isso é, análogos de açúcar contendo uma ligação dupla. A adição de reagente(s) apropriado(s) a essa ligação deve resultar na formação de ligações C-NH2 e C-OH nos dois átomos de carbono vizinhos, ligados por uma dupla ligação (Esquema 10).
Síntese de aminoaçúcares a partir de glicais
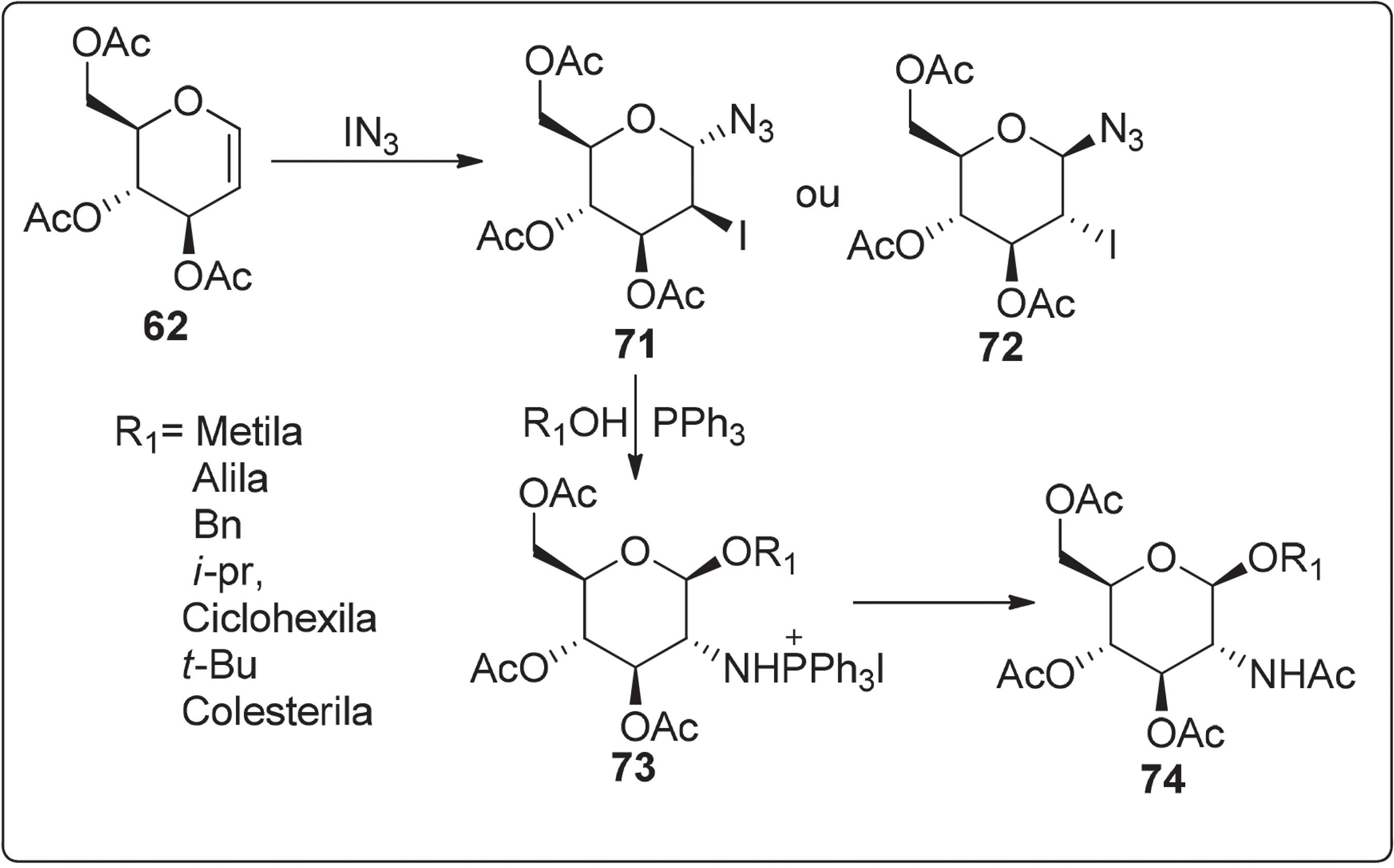
A síntese de aminoaçúcares foi realizada por Descotes e colaboradores,9292 Lafont, D.; Descotes, D.; Carbohydr. Res. 1987, 166, 195.,9393 Lafont, D.; Guilloux, P.; Descotes, D.; Carbohydr. Res. 1989, 61, 193. através da adição de iodoazida (IN3) ao tri-O-acetil-D-glical 62 fornencendo a mistura de 71 e 72 no qual o α-anômero 71 foi predominante (configuração D-mano). Os compostos 71 e 72 quando tratados separadamente com trifenilfosfina na presença de álcool, produziram sais de 2-aminofofôsnio de 1,2-trans-glicosídeos em bom rendimento. A remoção do grupo fofôsnio, sem isolamento do intermediário 73, seguido pela acetilação do grupo amino produziu o aminoaçúcar 74 (Esquema 11).
Dando continuidade, Lafont et al.9494 Lafont, D.; Woliny, A.; Boullanger, P.; Carbohydr. Res. 1998, 310, 9. demonstraram que os derivados de 6-amino-1,6-anidro-6-desoxiaçúcar (série D-glico, D-galacto, D-mano), podem ser preparados pelo tratamento de 1,2-trans-2-desoxi-2-iodo-β-D-glicopiranosilazida 72, com trifenilfosfina. O ataque nucleofilico do nitrogênio em C-2 produz uma aziridina intermediaria instável, que é aberta pelo álcool na posição anomérica.
Síntese de aminoaçúcares a partir de glicosídeos 2,3-insaturados
Em 2006, Mendlik e colaboradores9595 Mendlik, M. T; Tao, P.; Hadad, C. M.; Coleman, R. S.; Lowary, T. L.; J. Org. Chem. 2006, 71, 8059. descreveram um método de aziridinação fotoinduzida para a preparação da L-daunosamina e L-ristosamina. Na reação foi utilizado a L-treo-hex-2-enopiranosídeo 75 como material de partida para esta síntese (Esquema 12). O intermediário 76 sofreu uma irradiação com luz UV de 254 nm onde foi convertido em um derivado de aziridina 77. Para a abertura regiosseletiva do anel da aziridina 77, o mesmo foi submetido a uma reação de hidrogenação catalisada por Pd/C, seguido do tratamento com hidróxido de bário, originando o composto desejado 79 em rendimento de 87%.
Outro método usado para introdução da funcionalidade amino em glicosídeos 2,3-insaturados é a amina-hidroxilação catalisado por ósmio. O método consistiu em uma das etapas, sintetizar a oxazolidinona 84, como intermediário e em seguida a mesma é hidrolisada em uma solução aquosa de hidróxido de lítio para fornecer o composto 85 em rendimento de 96% e completa syn-diastereosseletividade. A síntese do metil-3-amino-3-desoxi-D-talopiranosídeo 85 é mostrada no Esquema 13.9696 Mirabella, S.; Cardona, F.; Goti, A.; Org. Lett. 2015, 17, 728.
Por outro lado, segundo os autores, a reação foi bem sucedida com o produto de partida 82, derivado do tri-O-acetil-D-galactal. Não obtiveram sucesso quando utilizaram o substrato, obtido a partir do tri-O-acetil-D-glical. Segundo eles, o resultado foi negativo quando se utilizou o tri-O-acetil-D-glical, porque de acordo com a conformação meia cadeira mais estável 5HO para o complexo intermediário 88 (Figura 4), observa-se um considerável impedimento estérico ao ataque das espécies de Os-imido com a parte aglicônica.
Conformação meia cadeira 5HO para o complexo intermediário resultante da reação de amina-hidroxilação com tri-O-acetil-D-glical 62
Mirabella e colaboradores9696 Mirabella, S.; Cardona, F.; Goti, A.; Org. Lett. 2015, 17, 728. também descreveram um protocolo de amina-hidroxilação dos derivados de hexenopiranosideo 87 protegidos (Esquema 14) – obtidos a partir de tri-O-acetil-D-glical 62, em cinco etapas e através de rearranjo Ferrier – e obtiveram o composto 88 em rendimento de 31%.
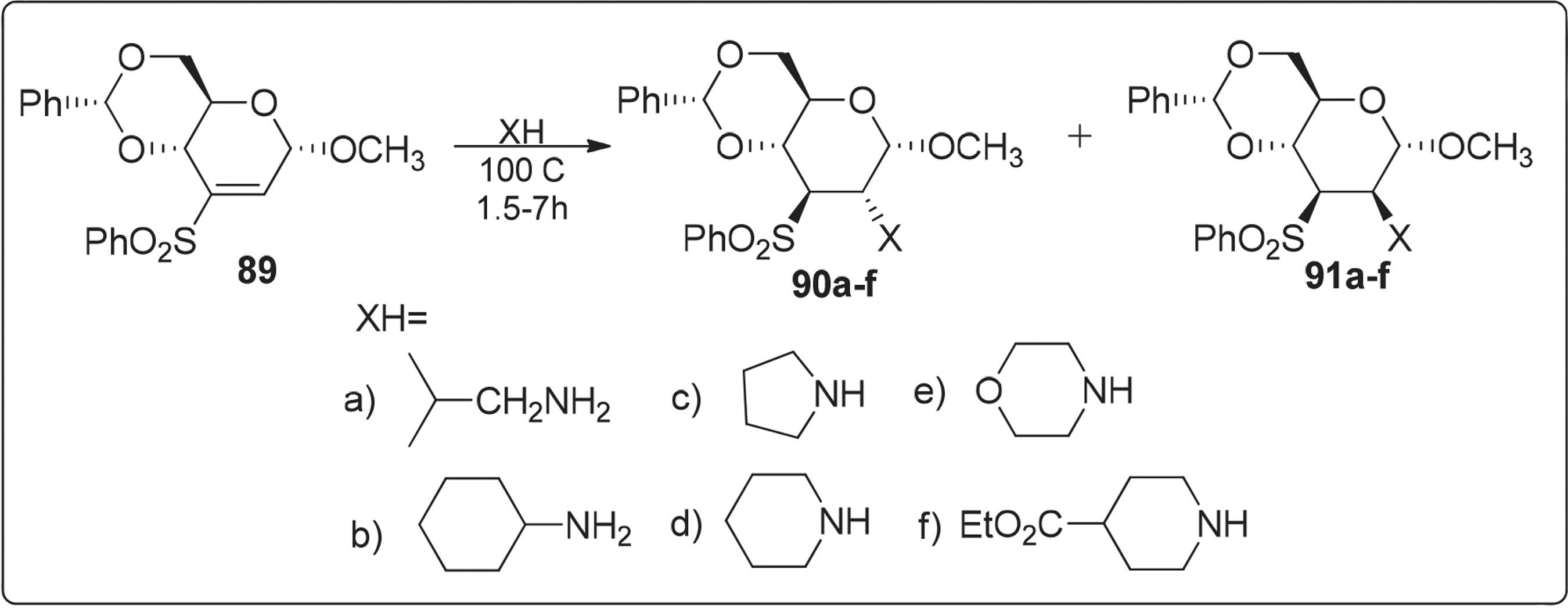
Ravindran e colaboradores9797 Ravindran, B.; Sakthivel, K.; Suresh, C. G.; Pathak, T.; J. Org. Chem. 2000, 65, 2637. relataram a síntese de uma nova classe de desoxiaminoaçúcares através da adição diastereoseletiva de aminas ao carboidrato modificado vinil sulfona. Essa metodologia de síntese consistiu em submeter o metil 2,3-didesoxi-4,6-O-(fenilmetileno)-3-C-fenilsulfonil-α-D-eritro-hex-2-enopiranosídeo 89 a uma reação de Michael com várias aminas, conforme Esquema 15.
Síntese de uma nova classe de desoxiaminoaçúcares através da adição diastereoseletiva de aminas
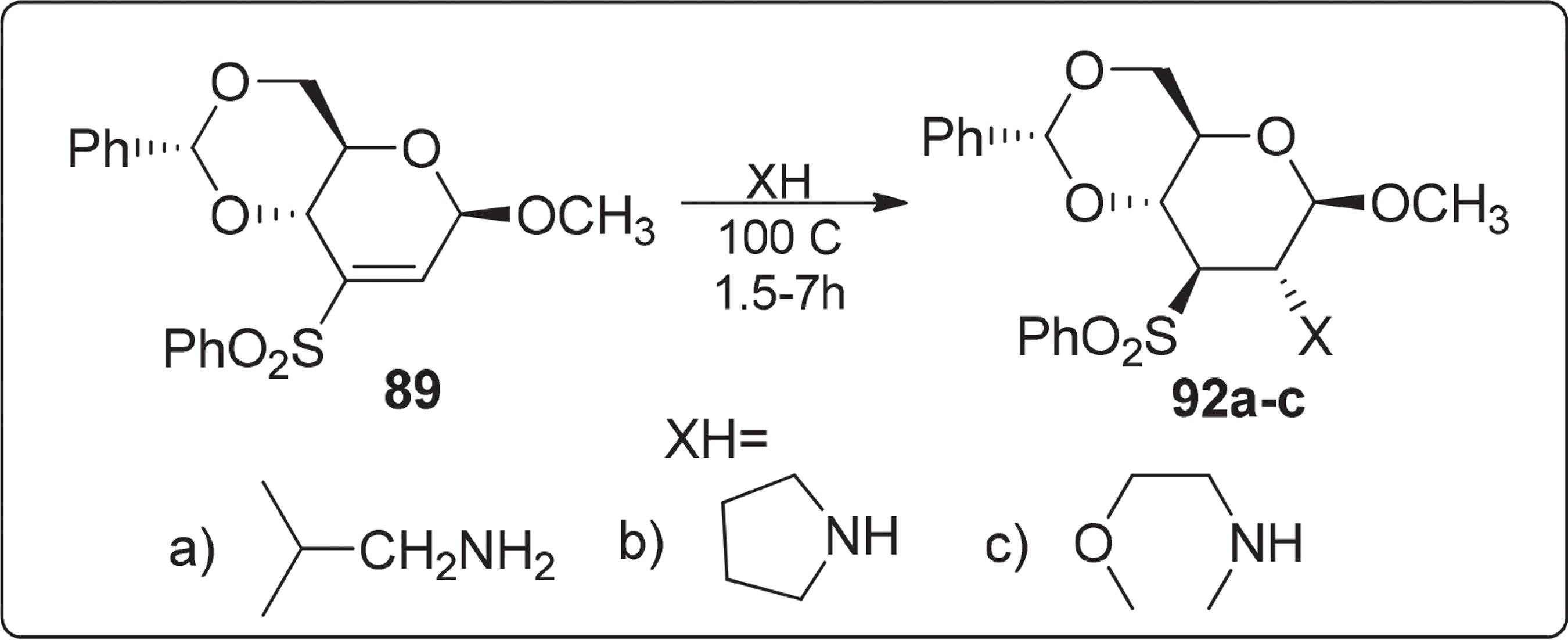
Por outro lado, os mesmos autores propuseram uma rota sintética onde o composto metil 2,3-didesoxi-4,6-O-(fenilmetileno)-3-C-fenil-sufonil-β-D-eritro-hex-2-enopiranosídeo 89 reagiu com várias aminas através de uma reação de Michael, Esquema 16. A rota de síntese de vários aminoaçúcares com configuração glico a partir de 89 constitui um novo método para introdução de amina N-monoalquiladas e N,N-dialquiladas ao carbono C-2 do anel piranosídico em configuração equatorial.
Síntese do metil 2,3-didesoxi-4,6-O-(fenilmetileno)-3-C-fenilsufonil-β-D-eritro-hex-2-enopiranosídeo 92a-c através de uma reação de Michael
Outra metodologia descrita na literatura9898 Oliveira, R. N.; Cottier, L.; Sinou, D.; Srivastava, R. M.; Tetrahedron 2005, 61, 8271.,9999 Zhang, G.; Shi, L.; Liu, Q.; Wang, J.; Li, L.; Liu, X.; Tetrahedron 2007, 63, 9705. consistiu no uso do catalisador paládio. Nessa metodologia foram utilizados nucleófilos contendo nitrogênio para incorporar a função azido em C-4 e C-2 em 2,3-didesoxihex-2-enopiranosídeos para obtenção dos aminoaçúcares 94a-c, 96a-c e 97a-c (Esquema 17).
Utilização de nucleófilos contendo nitrogênio para incorporar a função azido em C-4 e C-2 do 2,3-didesoxi hex-2-enopiranosídeos
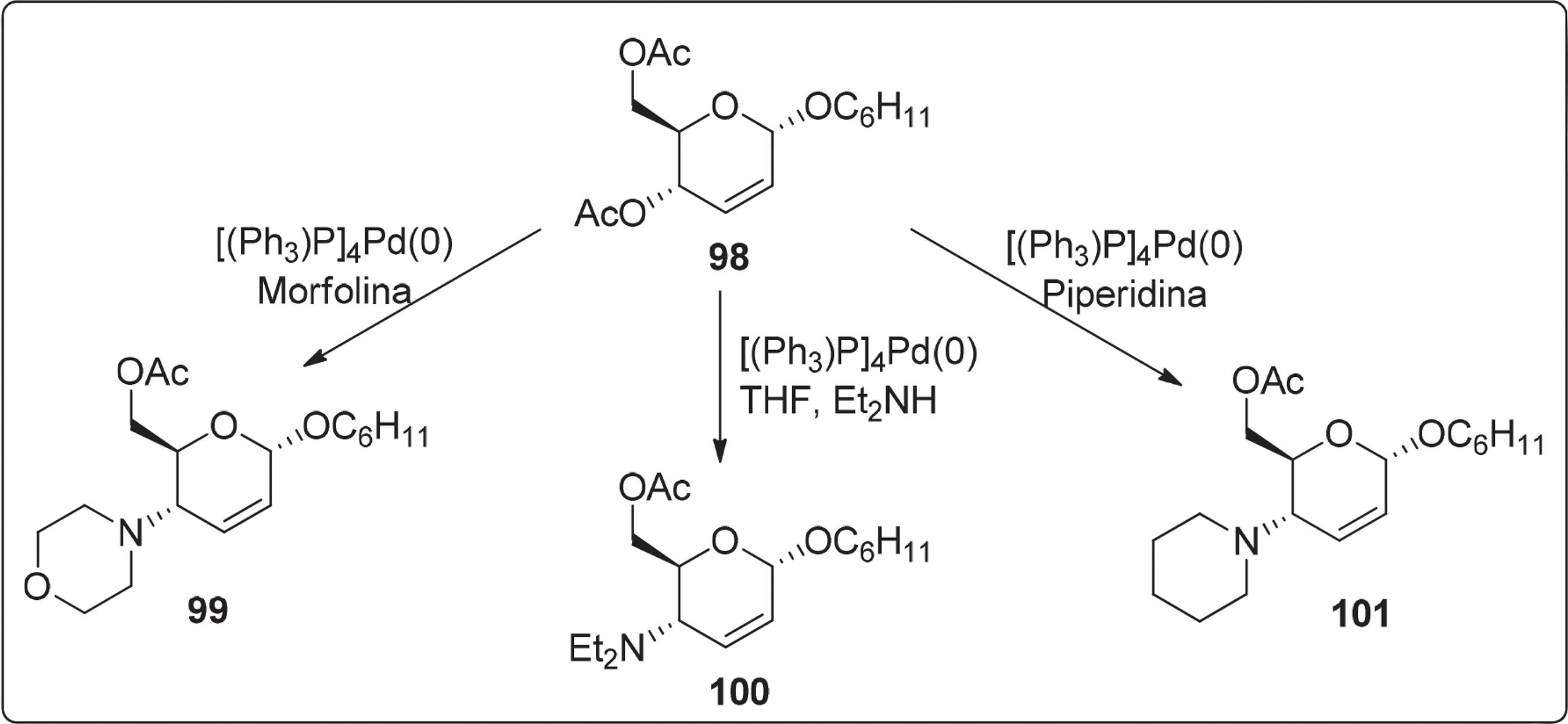
Há mais de 30 anos, Hanna e Baer100100 Baer, H. H.; Hanna, Z. S.; Can. J. Chem. 1981, 59, 889. propuseram a reação de aminação alilica, a qual consistia na síntese de aminoaçúcares insaturados a partir da transformação de anéis de piranoses insaturados na presença do catalisador paládio. Por outro lado, Brito e colaboradores101101 Brito, T. M. B.; Silva, L. P.; Siqueira, V. L.; Srivastava, R. M.; J. Carbohydr. Chem. 1999, 18, 609. utilizando o método descrito por Baer e Hanna desenvolveram a síntese de três novos 4-aminoaçúcares 2,3-insaturados 99-101 com rendimentos de 58-80% (Esquema 18) partindo de 4,6-di-O-acetil-2,3-didesoxi-α-D-eritro-hex-2-enopiranosídeo 98 e um catalisador de paládio zero. Posteriormente foi relatada a utilização de nucleófilos de nitrogênio envolvendo o uso de catalisadores de paládio e algumas fontes de amina.
A incorporação de grupos químicos que polariza a dupla ligação em O-glicosídeos 2,3-insaturados é possível através da Adição de Michael, que resulta em incorporação regiosseletiva do nucleófilo. Por exemplo, Mukherjee e Jayaraman102102 Mukherjee, A.; Jayaraman, N.; Carbohydr. Res. 2013, 380, 51. observaram que o composto 102 sofre adições nucleofílicas de grupos aminos em C-2, através de adição de Michael para fornecer o composto 103 e 104 com excelentes rendimentos e alta seletividade dependendo do nucleófilo e do substituinte de enxofre (Esquema 19). Outros exemplos de adição de Michael a dupla ligação de O-glicosídeos 2,3-insaturados são descritos na literatura.103103 Bussolo, V. D.; Romano. M. R.; Favero, L.; Pineschi, M.; Crott, P.; J. Org. Chem. 2006, 71, 1696.
Em 2002, Liberek et al.104104 Liberek, B.; Dabrowska, A.; Frankowski, R.; Matuszewska, M.; Smiatacz, Z.; Carbohydr. Res. 2002, 337, 1803. descreveram a síntese de 3-aminoaçúcares através da reação de adição de Michael a aldeídos insaturados derivados de tri-O-acetil-D-glical e tri-O-acetil-D-galactal, utilizando mercúrio (Hg2+) em dioxano, seguido da adição de azida de sódio e ácido acético levando a formação de 3-azido-2,3-dideoxiaçúcares na forma de diastereisômeros (Esquema 20). Os compostos obtidos foram submetidos à uma separação cromatográfica, uma vez que houve a formação da mistura de quatro diastereoisômeros, com o grupo azido preferencialmente na posição equatorial (eq / ax = 2,5:1). Após separados, os distereoisômeros 123 e 124 foram submetidos a uma reação de hidrogenação e acetilação resultando na porção acetilamino na posição C-3. Convém destacar que foi observada uma boa estereosseletividade na adição ao tri-O-acetil-D-glical e que a mesma foi perdida com a adição ao tri-O-acetil-D-galactal, isso provavelmente como consequência de impedimento estérico pelo grupo 4-acetoxi.105105 Ding, F.; William, R.; Wang, F.; Ma, J.; Ji, L.; Liu, X.-W.; Org. Lett. 2011, 13, 652.,106106 Ding, F.; William, R.; Wang, S.; Gorityala, B. K.; Liu, X.-W.; Org. Biomol. Chem. 2011, 9, 3929.
Síntese de aminoaçúcares de cadeia ramificada a partir de cetoaçúcares
Açúcares de cadeias ramificadas107107 Yoshimura , J.; Adv. Carbohydr. Chem. Biochem. 1984, 42, 69.,108108 Grisebach, H.; Schmid, R.; Angew. Chem., Int. Ed. 1992, 11, 159. e contendo nitrogênio ocorrem frequentemente como componentes de substâncias farmacologicamente ativas, muitos antibióticos pertencem a essa classe de compostos.109109 Hanessian, S.; Haskell, T. H. In The Carbohydrates; Pigman, W., Horton, D., eds.; Academic Press: New York, 1970, 139. Os cetoaçúcares α,β-insaturados são excelentes materiais de partida para a síntese de aminoaçúcares.110110 Brimacombe, J. S.; Angew. Chem., Int. Ed. 1992, 8, 401. Em 1982, Holder111111 Holder, N. L.; Chem. Rev. 1982, 82, 287. fez uma revisão destas hexenopiranosiduloses.
O primeiro exemplo de funcionalização de piranosídeo de alquila com cromóforo ceto α,β- insaturado foi realizado por Fraser-Reid e colaboradores112112 Fraser-Reid, B.; Mclean, H.; Usherwood, E. W.; J. Am. Chem. Soc. 1969, 91, 5392.,113113 Fraser-Reid, B.; Mclean, H.; Usherwood, E. W.; Yunker, M.; Can. J. Chem. 1970, 48, 2877. na síntese de alquil 2,3-didesoxi-hex-2-enopiranideos-4-ulose. A síntese de alguns desses derivados 2,3-didesoxi-4-ulose produz compostos estáveis e cristalinos. Em 1973, a síntese de alguns alquil 3,4-didesoxi-hex-2-enopiranosidulose foi relatada.114114 Holder, N. L.; Fraser-Reid, B.; Can. J. Chem. 1973, 51, 3357 e referências nelas citadas.,115115 Fraser-Reid, B.; Walker, D. L.; Ten, S. Y-K.; Holder, N. L.; Can. J. Chem. 1973, 51, 3950.
Reações de cicloadição
Algumas reações de cicloadição a cetoaçúcares foram propostas por Fraser-Reid e colaboradores.112112 Fraser-Reid, B.; Mclean, H.; Usherwood, E. W.; J. Am. Chem. Soc. 1969, 91, 5392.,113113 Fraser-Reid, B.; Mclean, H.; Usherwood, E. W.; Yunker, M.; Can. J. Chem. 1970, 48, 2877. Os autores obtiveram aminoaçúcares através de uma reação 1,3 dipolar de diazometano ao etil 6-O-acetil-2,3-dideosoxi-α-D-glicero-hex-2-enopiranosídeo-4-ulose 110 para formar o composto 111 (Esquema 21). A hidrogenação de 111 forneceu diamonoaçúcar ramificado 112, cuja configuração foi estabelecida pelo espectro de RMN 1H.116116 Srivastava, R. M.; Carthy, B. J.; Fraser-Reid, B.; Tetrahedron Lett. 1974, 2175.
Outra metodologia de síntese de aminoaçúcar a partir de enonas foi realizada por Apostolopoulos e colaboradores,117117 Apostolopoulos, C. D.; Couladouros, E. A.; Georgiadis, M. P.; Liebigs Ann. Chem. 1994, 781. pela adição de aminoácidos ao 5-C-substituído 2,3-didesoxi-hex-2-enopiranosid4-ulose 113, seguido de uma reação de redução para fornecer os compostos 114 (68%) e 115 (9%) com boas estereoseletividades (Esquema 22).
Aminoacúcares provenientes da adição de aminoácidos ao 5-C-substituído 2,3-didesoxi-hex-2-enopiranosid-4-ulose 113
Em 2003, Freitas Filho e colaboradores118118 Freitas Filho, J. R.; Srivastava, R. M.; Silva, W. J. P.; Cottier, L.; Sinou, D.; Carbohydr. Res. 2003, 338, 673. descreveram a reação de cicloadição 1,3-dipolar de nitrona a hexenulose, que consistiu em reagir as enonas 116a-c, com N-fenilnitrona 117 resultando na formação dos compostos 118a-c em rendimentos variando de 52-76% (Esquema 23).
Convém destacar que os autores do trabalho propuseram um mecanismo que consistiu na adição de óxido de metilidenoanilina 117 a dupla ligação C2-C3 de 116a-c em face oposta a aglicona dando uma isoxazolidina, via estado de transição 119, conforme Esquema 24.
Outros exemplos de reação 1,3-dipolar para a síntese de aminoaçúcares foi relatado por Dahl e colaboradores (Esquema 25).119119 Dahl, R. S.; Finney, N. S.; J. Am. Chem. Soc. 2004, 126, 8356.,120120 Dahl, R. S.; Baldridge, K. K.; Finney, N. S.; Synthesis 2010, 2292. Nessa síntese o galactal 120 reagiu com azidas benzílicas em trietilortoformato como solvente para fornecer o intermediário triazolina 121, o qual por irradiação forneceu a N-benzil aziridina 122. Segundo os autores, o tratamento adicional da reação com uma base forte proporcionou formação de um derivado de aminoglicosídeo. Tal cicloadição dipolar pode ser usada para obter 2-amino-2-desoxiglicosídeo 123.
A literatura também cita a síntese de precursores de aminoaçúcares através da adição 1,3-dipolar de diazometano a dupla ligação de hexenulose121121 Srivastava, R. M.; Souza, A. M. A.; Silva, L. P.; Freitas Filho, J. R.; Hallwass, F.; J. Braz. Chem. Soc. 2002, 13, 158. e cicloadição 1,3-dipolar de fenilazida a diferentes hexenuloses.122122 Freitas Filho, J. R.; Cottier, L.; Srivastava, R. M.; Sinou, D.; Synlett 2003, 1358.
Síntese de aminoaçúcares a partir de outras rotas sintéticas
Ji e colaboradores123123 Ji, X-M.; Mo, J.; Liu, H-M.; Sun, H-P.; Carbohydr. Res. 2006, 341, 2312. relataram a síntese de diversos aminoacúcares derivados a partir de cetoaçúcares proveniente da D-xilose através de uma série de reações, por exemplos a reação de Henry, reações de hidrogenação e reações de adição nucleofílica ou reações de substituição. Segundo os autores em presença de KF, a adição de nitrometano ao grupo carbonila de 5-O-benzoil-1,2-O-isopropilideno-D-eritro-cetofurano-3-ulose 124 ocorreu estereosseletivamente para fornecer apenas o isômero 125 com configuração ribo e rendimento de 95% (Esquema 26). A presença de um grupo nitro na estrutura foi confirmada por análise de infravermelho e a configuração absoluta de todos os átomos de carbono assimétricos foi determinada pela análise de difração de raios-X. A estereoseletividade provavelmente resultou no impedimento estérico do grupo 1,2-O-isopropilideno. A etapa seguinte consistiu na hidrogenação catalítica de 125 sob condições neutras, que levou a uma mistura dos compostos 126a e 126b, os quais foram isolados por cromatografia em coluna um sistema de MeOH-CHCl3 1:15 para fornecer o composto desejado 126a em 75% de rendimento e o composto 126b, com 10% de rendimento. A estrutura de 126a foi confirmada pelos espectros 1H, 13C, RMN 2D, HRMS e por cristalografia de raios-X.
Recentemente, Zhu et al.124124 Zhu, Z.; Glazier, D. A.; Yang, D.; Tanga, W.; Adv. Synth. Catal. 2018, 360, 2211. relataram a síntese assimétrica catalítica de oito possíveis 2,3,4,6-tetradesoxi-4-amino-hexopiranosídeos, dentre eles os compostos 129, 132, 135 e 138 (Esquema 27), e também a síntese das gliconas dos produtos naturais grecociclina A e B, espinosina A e ossamicina. Entretanto, segundo os autores, a diastereosseletividade para um dos três centros estereogênicos foi baixa e não controlada por catalisadores.
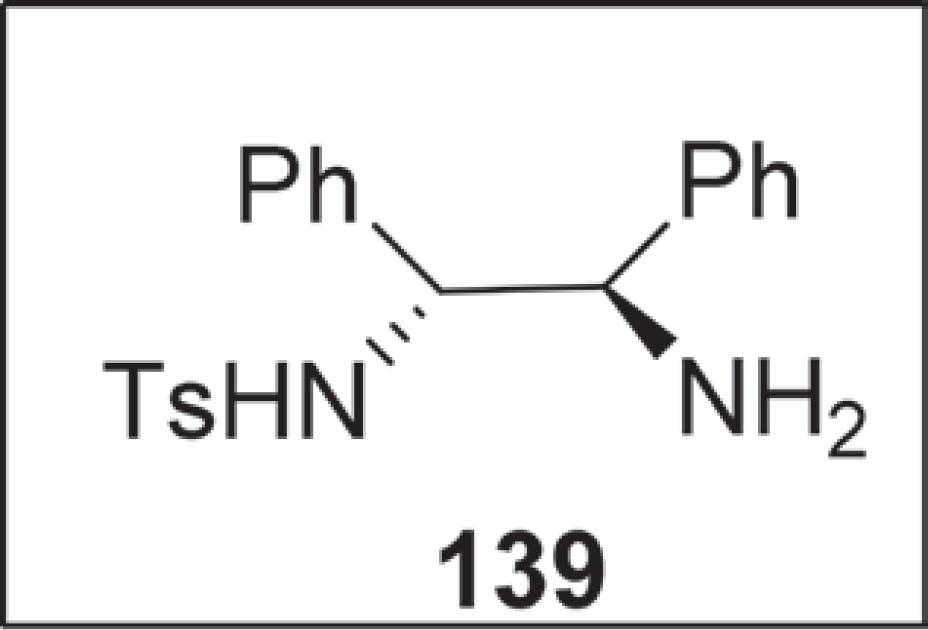
Convém destacar que uma das etapas de síntese de 2,3,4,6-tetradesoxi-4-amino-hexopiranosídeos foi a partir do lactol opticamente puro (98% ee), o qual foi preparado em duas etapas via redução catalítica assimétrica do 2-acetilfurano mediada por [Cp*RhC]2/(R,R)-Ts-DPEN 139 (Figura 5) seguido por um rearranjo de Achmatowicz.125125 Ding, W.; Yu, J.-P.; Shi, X.-X.; Nie, L.-D.; Quan, N.; Li, F.-L. Tetrahedron: Asymmetry 2015, 26, 1037.
Em 2015, Ding e colaboradores126126 Song, W.; Zhao, Y.; Lynch, J. C.; Kim, H.; Tang, W.; Chem. Commun. 2015, 51, 17475. descrevem a síntese estereosseletiva do pseudo-aminoaçúcar (+)-valienamina 144 a partir do ácido (-)-chiquímico 140 com rendimento global de 38,3% através de 13 etapas reacionais (Esquema 28). Entre essas etapas reacionais, destaque é dado à azidação através de uma substituição nucleofílica do tipo SN2 do grupo Oms do C-5 com azida de sódio e cloridrato de trietilamina, proporcionando o composto 142 com rendimento de 90%. A configuração (R) de C-5 foi invertida para a configuração (S) através de uma inversão do tipo Walden. Após algumas etapas reacionais, foi promovida a hidrogenação altamente seletiva do grupo azido (N3), transformando-o no grupo amina, utilizando o catalisador de Lindlar e convertendo na (+)-valienamina 144 com rendimento de 91%.
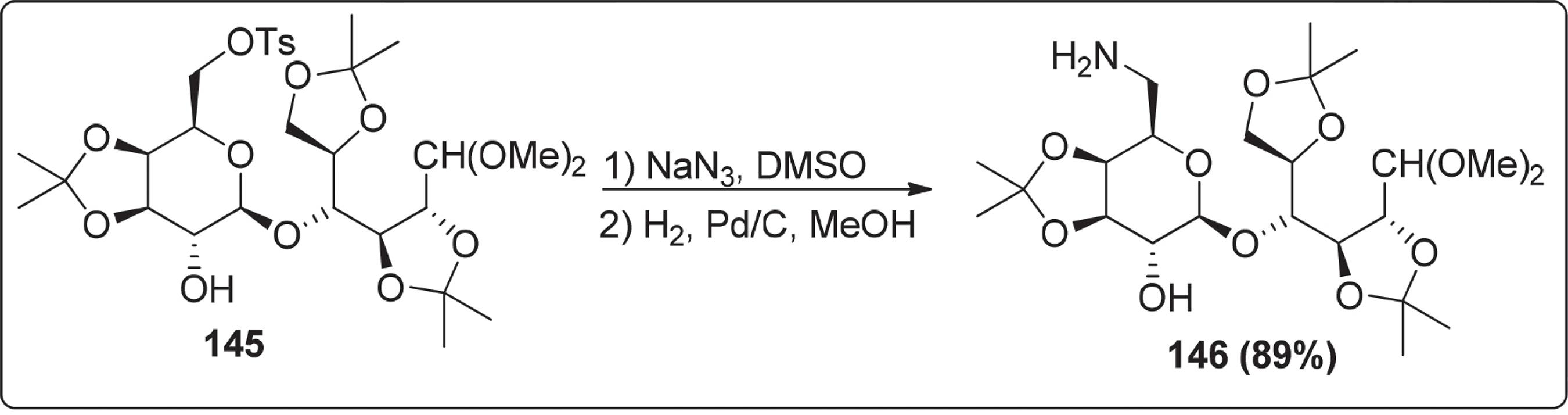
Em 2015, Corsi et al.127127 Corsi, M.; Bonanni, M.;Catelani, G.; D’Andrea, F.; Gragnani, T.; Bianchini, R.; J. Org. Chem. 2013, 3, 41. descreveram a síntese de derivados de aminoaçucar via intermediário de N-benzilamina, através da substituição do grupo tosilato por azida Nessa síntese os autores utilizaram solventes apróticos polares como N,N-dimetilformamida (DMF) ou dimetilsulfóxido (DMSO) e obtiveram o composto 146 em rendimento de 89% (Esquema 29).
Outras estratégias sintéticas que têm sido empregadas na síntese de aminoaçúcares, incluindo reação de substituição em vários átomos de carbono do anel piranosídico ou furanosídico, são bem descritas em revisão publicada no ano de 2016 e em artigos nela citada.88 Skarbek, K.; Milewska, M. J.; Carbohydr. Res. 2016, 434, 44.
APLICAÇÕES DE AMINOACÚCARES
Na Medicina
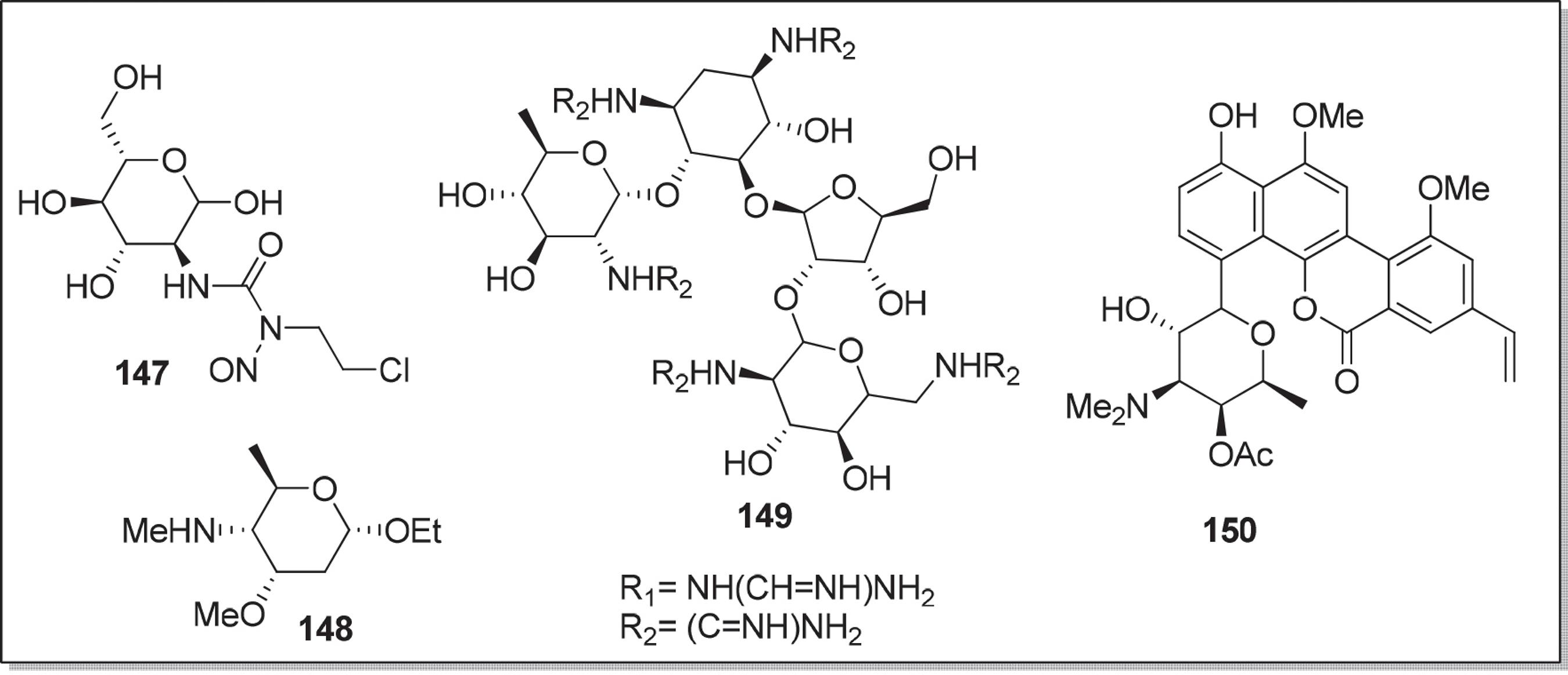
Assim como os açúcares de cadeia ramificada e cetoaçúcares, os aminoaçúcares tem aplicações farmacológicas, pois alguns possuem atividade antitumoral como a clorozoticina 147.128128 Suami, T.; Machinami, T.; Hitsamatsu, H.; J. Med. Chem. 1979, 22, 247. Já a holocosamina 148 (Figura 6) apresenta atividades antifúgica129129 Omura, S.; Katagiri, M.; Atsumi, K.; Hata, T.; J. Chem. Soc Perkin 1, 1974, 0, 1627. e cardiotônica.130130 Georges, M.; Mackay, D.; Fraser-Reid, B.; Can. J. Chem. 1984, 62, 1539. Todavia, a guanidioneomicina 149 possui atividade anti-HIV. Futagami e colaboradores131131 Futagami, S.; Ohashi, Y.; Imura, K.; Hosoya, T.; Ohmori, K.; Matsumoto, T.; Suzuki, K.; Tetrahedron Lett. 2000, 41, 1063. demostraram em seu trabalho a síntese total da ravidomicina 150, um aminoaçúcar antibiótico com atividade antitumoral.
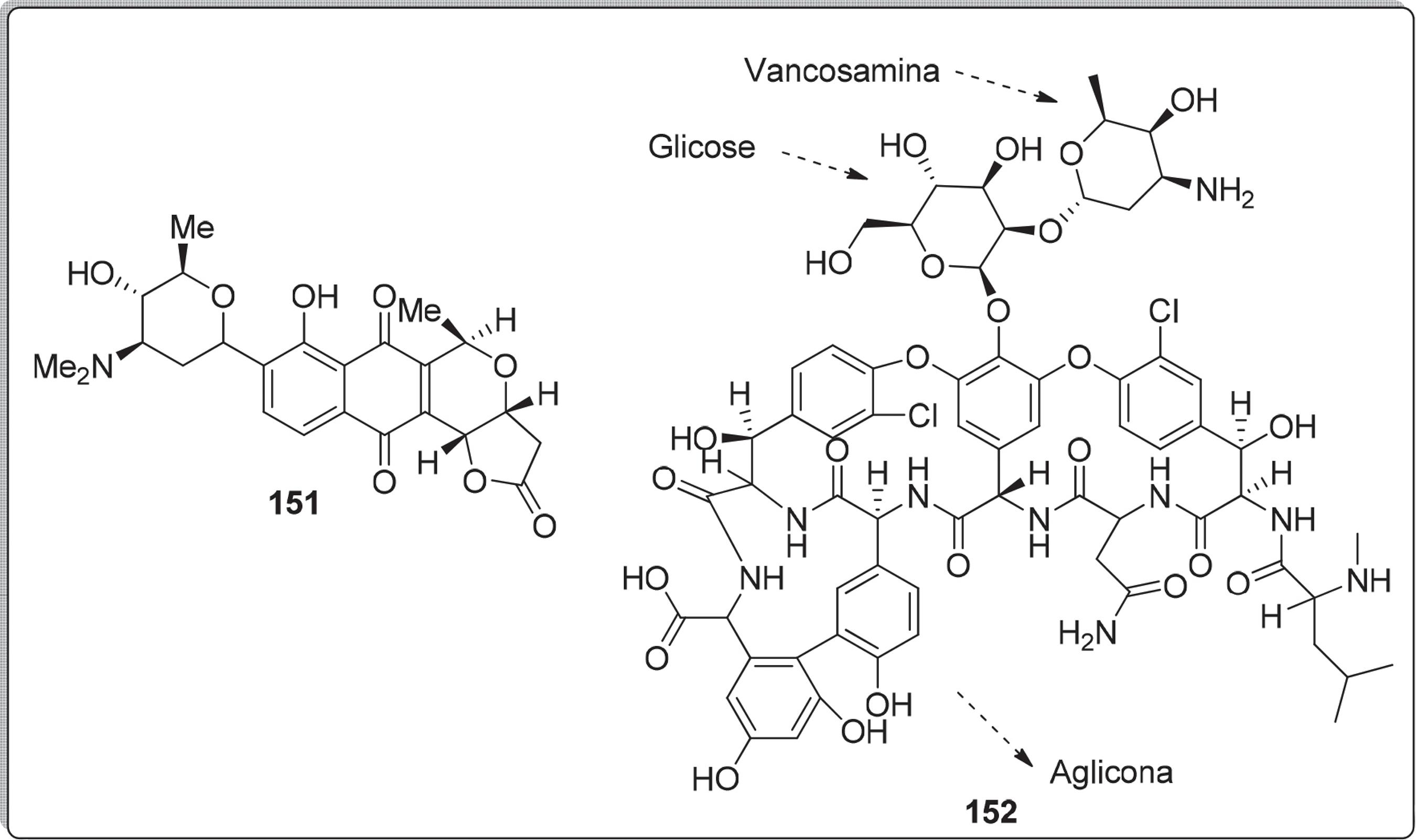
A Figura 7 apresenta outros exemplos de compostos que contém aminoaçúcares nas estruturas, tais como, medermicina 151132132 Brimbe, M. A.; Brenstrum, T, J.; Tetrahedron Lett. 2000, 41, 1107. e vancomicina 152.133133 Johnson, A. W.; Smith, R. M.; Guthrie, R. D.; J. Chem. Soc. Perkin Trans 1. 1972, 2153.
134 Smith, G. R.; Giuliano, R. M.; Carbohydr. Res. 2000, 323, 208.
135 Willians, D. H.; Bardsley, B.; Angew. Chem., Int. Ed.
1999, 28, 1172.-136136 Fu, X.; Albermann, C.; Jiang, J.; Liao, J.; Zhang, C.; Thorson, J.; Nat. Biotechnology
2003, 21, 1467. O antibiótico medermicina tem aplicação na síntese de biomoléculas e na agregação plaquetária. A vancomicina, um antibiótico glicopeptídeo contendo o açúcar L-vancosamina como unidade de aminoaçúcar, tem aplicação no tratamento de infecções por bactérias gram-positivas resistentes a meticilina. Os aminoaçúcares são ainda utilizados também na síntese de oligossacarídeos.137137 Banoub, J.; Boullanger, P.; Lafont, D.; Chem. Rev.
1992, 92, 1167.
A necessidade da descoberta de novos antibióticos é cada vez mais intensa, dado que, conforme descrito na literatura, as bactérias adquirem resistências aos mesmos. Na literatura podem ser encontrados relatos de vários compostos e seus análogos, que contém aminoaçúcares, tais como tobamicina, acarbose e salbostatina.138138 Coben, M. L.; Science
1992, 257, 1050.
139 Neu, H. C.; Science
1992, 257, 1064.
140 Lin, H.; Walsh, C. T.; J. Am. Chem. Soc.
2004, 126, 13998.-141141 Chang, C.-W. T.; Hui, Y.; Elchert, B.; Wang, J.; Li, J.; Rai, R.; Org. Lett.
2002, 4, 4603. Dentre a classe de antibióticos dos aminoglicosídeos, os 4-aminoaçúcares vem sendo bastante estudados, tais como as piramicinas,141141 Chang, C.-W. T.; Hui, Y.; Elchert, B.; Wang, J.; Li, J.; Rai, R.; Org. Lett.
2002, 4, 4603.,142142 Elchert, B.; Li, J.; Wang, J.; Hui, Y.; Rai, R., Ptak, R.; Ward, P.; Takemoto, J. Y.; Bensaci, M.; Chang, C. -W. T.; J. Org. Chem. 2004, 69, 1513. as aprimicinas143143 O’Connor, S.; Lam, L. K. T.; Jones, N. D.; Chaney, M. O.; J. Org. Chem. 1976, 41, 2087. e apicamicina.144144 S uzuki, T.; Suzuki, S. T.; Yamada, I.; Koashi, Y.; Yamanda, K.; Chida, N.; J. Org. Chem. 2002, 67, 2874.
Convém destacar que outras aplicações de aminoaçúcares na medicina são como inibidores de glicosidases, como exemplos podemos citar a acarbose e salbostatina.145145 McAuliffe, J. C.; Stick, R. V.; Stone, B. A.; Tetrahedron Lett. 1996, 37, 2479.
Os exemplos de aminoaçúcares descrito nos parágrafos anteriores, com potentes atividades farmacológicas, são relativamente poucos em comparação com os existentes e que mereciam ser citados neste trabalho.
Na agricultura
Os aminoaçúcares são utilizados como biomarcadores na agricultura, pois esses compostos fazem parte da parede celular de bactérias, fungos e actinomicetos.146146 Amelung, W. In Assessment Methods for Soil Carbon; Lal, R., Kimble, J. M., Follett, R. F., Stewart, B. A., eds.; Lewis Publishers: Boca Raton, 2001, pp. 233-272.
147 Hu , Y.; Zheng, Q.; Zhang, S.; Noll, L.; Wanek, W.; Soil Biol. Biochem.
2018, 123 , 115.-148148 Kandeler, E.; Tscherko, D.; Bruce, K. D.; Stemmer, M.; Hobbs, P. J.; Bardgett, R. D.; Amelung, W.; Biol. Fertil. Soils
2000, 32, 390. As paredes celulares bacterianas contêm um peptidoglicano, construído a partir dos derivados da D-glicose, N-acetilglicosamina e ácido N-acetilmurâmico. A estrutura do peptidoglicano está presente apenas em células procariotas e nunca foi encontrada em células eucarióticas. Como biomarcadores, eles são usados para rastrear resíduos bacterianos, fúngicos e actinomicetos que contribuem para formação da matéria orgânica do solo diante da presença dos aminoaçúcares glicosamina, galactosamina e N-acetilmurâmico 153 (Figura 8). As relações glicosamina/ácido murâmico (Glic/Mur.) e glicosamina/galactosamina (Glic/Gal) podem servir como indicadores da contribuição relativa fúngica e bacteriana para a MOS.
Em bactérias gram-positivas, 90% da parede celular consistem em peptidoglicano, enquanto em bactérias gram-negativas, esse número varia de 5-20%.149149 Brock, T. D.; Madigan, M. T.; Biology of Microorganisms, Prentice-Hall: Englewood Cliffs,1988. Acredita-se que os fungos contenham apenas glicosamina e galactosamina.150150 Cochran, T. W., Vercellotti, J. R.; Carbohydr. Res. 1978, 61, 529. Além disso, os exoesqueletos de invertebrados contêm quitina, um polímero de N-acetilglicosamina.151151 Chantigny, M. H.; Angers, D. A.; Prevost, D.; Vezina, L. P.; Chalifour, F. P.; Soil Sci. Soc. Am. J. 1997, 61, 262. Sabe-se que a glicosamina foi encontrada no revestimento intestinal de minhocas, nas cascas dos ovos dos nematoides, nos polissacarídeos dos moluscos e na gelatina do caracol.152152 Zhanga, X.; Amelunga, W.; Yuanb, Y.; Samson-Liebigc, S.; Brown, L.; Zecha, W.; Appl. Soil Ecol.
1999, 11, 271. No entanto, as concentrações de aminoaçúcares e ácido murâmico são aplicadas rotineiramente para indicar contribuições microbianas à matéria orgânica do solo (MOS).153153 Solomon, D.; Lehmann, J.; Zech, W.; Biol. Fertil. Soils
2001, 33, 85.
154 Turrión, M.-B.; Glaser, B.; Zech, W.; Biol. Fertil. Soils
2002, 35, 49.-155155 Amelung, W.; Zhang, X.; Flach, K.W.; Zech, W.; Soil Sci. Soc. Am. J. 1999, 63, 86.
Glaser e colaboradores156156 Glaser, B.; Turrión, M-B.; Alef, K.; Soil Biol. Biochem. 2004, 36, 399. investigaram o padrão, quantidade e dinâmica de três aminoaçúcares (glicosamina, manosamina e galactosamina) e ácido murâmico na biomassa microbiana total de bactérias, fungos e actinomicetos seletivamente cultivados de cinco solos diferentes, com e sem glicose.
Na indústria de polímeros e têxteis
A incorporação de unidades derivadas de açúcar em polímeros tradicionais de crescimento em etapas, tais como poliamidas e poliésteres, é potencialmente um método de interesse para preparar novos materiais biodegradáveis e biocompatíveis para aplicação em produtos biomédicos e outros setores de maior consumo, como embalagens de alimentos.157157 Yokoe, M.; Aoi, M.; Okada, M.; J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 3909.,158158 García-Martín, M. G.; Pérez, R. R; Hernández, E. B.; Espartero, J. L.; Muñoz-Guerra, S.; Galbis, J. A.; Macromolecules
2005, 38, 8664. As principais razões para esse interesse são a grande abundância de açúcares naturais, sua diversidade estrutural, suas múltiplas funcionalidades e a sua natureza hidrofílica dos materiais resultantes, resultando em maior degradabilidade hidrolítica.159159 Göpferich, A.; Biomaterials
1996, 17, 103. Além disso, seu impacto ambiental é menor que o dos polímeros clássicos.160160 Cunliffe, D.; Pennadam, S.; Alexander, C.; Eur. Polym. J.
2004, 40, 5. Entretanto, embora os polímeros tenham sido sintetizados usando monômeros derivados de açúcar com grupos hidroxila livres,161161 Kiely, D. E.; Chen, L.; Lin, T. H.; J. Am. Chem. Soc.
1994, 116, 571.
162 Kiely, D. E.; Chen, L.; Lin, T. H.; J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 594.-163163 Styron, S. D.; Kiely, D. E.; Ponder, G.; J. Carbohydr. Chem.
2003, 22, 123. a maioria das sínteses de polímeros lineares de alto peso molecular envolve derivados que possuem grupos hidroxila adequadamente protegidos.164164 Galbis, J. A.; García-Martín, M. G. In Monomers, polymers and composites from renewable resources; Belgacem, M. N., Gandini, A., eds.; Elsevier: Oxford., 2008, cap. 5.
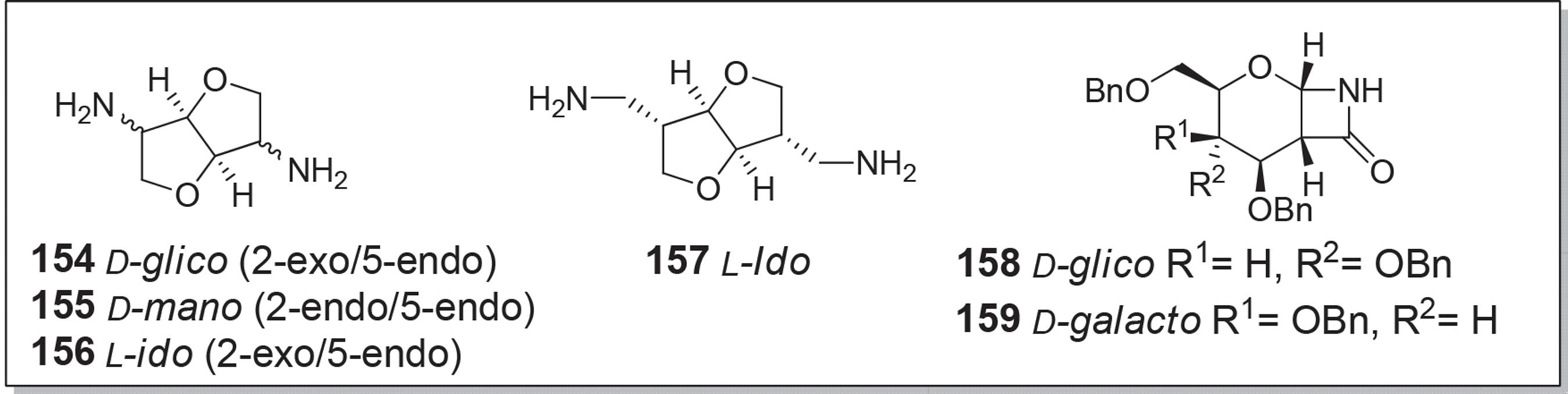
Logo, vários monômeros de aminoaçúcares (154-159) têm sido amplamente utilizados para preparar poliamidas, poliuretanos e poliureias (Figura 9).165165 Galbis, J. A.; Martín, M. de G. G. M.; Paz M. V.; Galbis. E.; Chem. Rev. 2016, 116, 1600.
Devido às boas propriedades térmicas e mecânicas, as poliamidas do tipo Nylo constituem um dos grupos mais importantes de polímeros de condensação e são amplamente utilizadas na indústria para moldagem por injeção e aplicações de filme ou fibra. Assim, a preparação de poliamidas do tipo Nylon mais hidrofílicas e degradáveis a partir de monômeros a base de açúcar representa um grande desafio. As primeiras sínteses de poliamidas à base de açúcar produziram apenas fibras frágeis e de baixo peso molecular. Os monômeros de açúcar necessários para a síntese de poliamidas são diaminossacarídeos, ácidos aldáricos ou ácidos aminoaldônicos. A introdução de grupo amino é geralmente realizada através de um éster sulfonato, a partir de um deslocamento do tipo SN2 por azida, seguido de uma reação de hidrogenação.166166 Martín, M. G.; Perez, R. R.; Hernández, E. B.; Galbis, J. A.; Carbohydr. Res. 2001, 333, 95.
Koning et al.167167 Jasinska, L.; Villani, M.; Wu, J.; Es, V. D.; Klop, E.; Rastogi, S.; Koning, C. E.; Macromolecules 2011, 44, 3458. descreveram homo- e copoliamidas totalmente de base biológica a partir de ácido sebácico, 2,5-diamino-2,5-didesoxi-1,4,3,6-dianidroiditol (diaminoisoidida, 156) e 1,4-diaminobutano. Poliamidas de baixo peso molecular foram obtidas por poli-condensação dos sais à base desses monômeros ou por poli-condensação interfacial. Poliamidas de maior peso molecular foram obtidas por polimerização em estado sólido (SSP) dos pré-polímeros. As técnicas de FT-IR e raios-X foram utilizados para a investigação da estrutura cristalina dos polímeros após o SSP. Conformação local e co-cristalização dessas poliamidas à base de diaminoisoidídeos foram estudadas por FT-IR, RMN de estado sólido e WAXD.168168 Walc, L. J.; Villani, M.; Dudenko, D.; Asselen, O. V.; Klop, E.; Rastogi, S.; Hansen, M. R.; Koning, C. E.; Macromolecules 2012, 45, 2796.
Os poliuretanos têm sido extensivamente estudados nas últimas décadas, e têm sido objetos de inúmeras patentes, documentos e livros. Eles são usados principalmente como materiais de mercadoria e em aplicações industriais. No entanto, como alguns deles são biodegradáveis e biocompatíveis, e seu uso em aplicações médicas está sendo amplamente investigado devido à baixa toxicidade, biodegradabilidade potencial, biocompatibilidade e estruturas versáteis, que os tornam adequados como parte de sistemas de administração de medicamentos, como curativos dermatológicos, como materiais hemocompatíveis para cateteres e como filtros em instrumentação e dispositivos biomédicos.
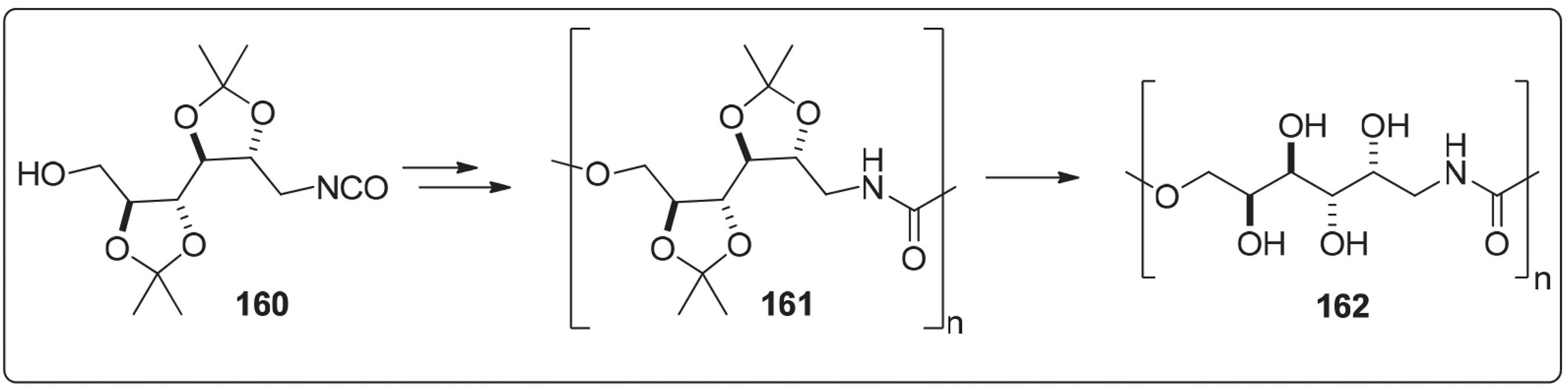
Gómez e Varela169169 Gomez, R. V.; Varela, O.; Macromolecules 2009, 42, 8112. descreveram a síntese de um [AB]-poliuretano estereorregular 162 a partir do 1-desoxi-1-isocianato-2,3:4,5-di-O-isopropilideno-D-galactitol 160. Segundo os autores a hidrólise dos grupos isopropilideno de 161 forneceu o polihidroxi [n]-poliuretano 162 (Esquema 30); no entanto, os estudos de degradação hidrolítica não foram realizados.
Síntese de um [AB]-poliuretano estereorregular a partir de 1-desoxi-1-isocianato-2,3:4,5-di-O-isopropilideno-D-galactitol
Embora, segundo Lu et al.,170170 Lu, H.; Sun, P.; Zheng, Z.; Yao, X.; Wang, X.; Chang, F. C.; Polym. Degrad. Stab. 2012, 97, 661. algumas poli (uretano-ureia) degradáveis baseadas em recursos renováveis – como a L-cistina – tenham despertado certo interesse, porque poderiam potencialmente ser aplicadas como biomateriais temporários, as poliureias à base de açúcar atraíram menos atenção até hoje. Thiem et al.171171 Bachmann, F.; Reimer, J.; Ruppenstein, M.; Thiem, J.; Macromol. Chem. Phys. 2001, 202, 3410. descreveram a preparação de poliureias à base de açúcar: inicialmente, o diamino dihidrocloreto (165-167) foi reagido com fosgênio em tolueno (Esquema 31) para fornecer os diisocianatos (166-170) em bons rendimentos. Em seguida, através de uma reação de poliadição, os polímeros 169-171 são obtidos em bons rendimentos.
CONSIDERAÇÕES FINAIS
O crescente interesse na síntese, biossíntese, ocorrência natural e aplicações de aminoaçúcares tem estimulados diversos pesquisadores na área da química dos carboidratos a respeito do papel de alguns destes compostos de interesse biológico devido à ocorrência em muitas substâncias biologicamente ativas, especialmente os antibióticos, tais como gentamicina, neomicina, estreptomicina, kanamicina, dentre outros. Por outro lado, quando uma molécula de aminoaçúcar está presente na estrutura de um antibiótico, esse supera seu espectro de atividade, melhorando sua atividade e os perfis físico-químico e farmacocinético.
Os aminoaçúcares podem ser obtidos através de duas rotas biossintéticos. Nessas rotas os aminoaçúcares são derivados do metabolismo primário e metabolismo secundário, que consiste na conversão de um monossacarídeo – geralmente na forma de açúcar fosfatado ou de nucleotídeo glicosilado – catalisada por uma enzima aminotransferase específica ou uma amidotransferase. Ao contrário da biossíntese de aminoacúcares no metabolismo primário, que deriva da aminação catalisada pela D-glucosamina-6P sintase de D-frutose-6P, a biossíntese de aminoacúcares no metabolismo secundário envolve a introdução da funcionalidade amino por uma enzima aminotransferase PLP-dependente. Os aminoacúcares derivados de metabólitos secundários são cruciais para as atividades biológicas ideais dos compostos de origem.
Algumas das estratégias sintéticas para obtenção de aminoaçúcares envolvem reações de substituição em vários átomos de carbono do anel piranosídico ou furanosídico através da: a) clivagem de derivados epoxi-açúcares por aminas; b) redução de derivados de oxima e azida; c) adição a ligações duplas de glicais e de glicosídeos 2,3-insaturados e d) cicloadição à cetoaçúcares, dentre outras. Algumas dessas estratégias não fornecem acesso a aminoaçúcares com regio- e estereoseletividade completas, uma vez que, em muitos casos, são necessárias separações difíceis das misturas formadas e, em consequência, os rendimentos finais são baixos.
As primeiras estratégias de síntese de aminoaçúcares foram baseadas em monossacarídeos facilmente disponíveis como precursores, tendo a configuração nos estereocentros do açúcar bem definidas. A maioria dos métodos modernos de síntese de aminoaçúcares que consiste em incorporar um grupo amino ligado ao carbono 2, 3, 4 e 6 envolve glicais ou galactal como produto de partida, que são substratos relativamente baratos e comercialmente disponíveis. A presença de centros estereogênicos no esqueleto do D-glical ou do D-galactal pode ser explorada para introduzir novas funcionalidades de maneira estereosseletiva.
Esta revisão aborda as principais reações de sínteses e transformações enzimáticas na síntese de aminoacúcares, bem como a sua reatividade química, com foco particular nos desenvolvimentos ocorridos nos últimos anos. Por fim, estamos convencidos de que os resultados aqui descritos poderão ser úteis para pesquisadores que desejam utilizar a química dos carboidratos, mais especificamente a química dos aminoaçúcares e suas aplicações biológicas como objeto de estudo.
AGRADECIMENTOS
Os autores agradecem à Capes e à FACEPE pelo apoio recebido.
REFERÊNCIAS
-
1Teranishi, K.; Carbohydr. Res 2002, 337, 613.
-
2Liu, F. W.; Zhang, Y. B.; Liu, H. M.; Song, X. P.; Carbohydr. Res 2005, 340, 489.
-
3Taylor, C. M.; Tetrahedron 1998, 54, 11317.
-
4Tu, D.; Blaha, G.; Moore, P. B.;. Steitz, T. A.; Cell 2005, 121, 257.
-
5Mankin,A. S.; Curr. Opin. Microbiol. 2008, 11, 414.
-
6Zhang, Z.; Fukuzaki, T.; Myer, A. G.; Angew. Chem., Int. Ed. 2016, 55, 523.
-
7Zhu, R.; Lin, Y.-S.; Lipp, J. S.; Meador, T. B.; Hinrichs, K.-U.; Biogeosciences 2014, 11, 4869.
-
8Skarbek, K.; Milewska, M. J.; Carbohydr. Res 2016, 434, 44.
-
9Fischer, E.; Leuchs, H.; J. Chem Pub Soc. Eur 1902, 35, 3787.
-
10El Ashry, E. S. H.; Aly, M. R. E. P.; Pure Appl. Chem. 2007, 79, 2229.
-
11Arsequell, G.; Valencia, G.; Tetrahedron: Asymmetry 1999, 10, 3045.
-
12De Freitas Filho, J. R.; Srivastava, R. M.; Carbohydr. Res 2003, 338, 673.
-
13Salton, M. R. J.; Annu. Rev. Biochem 1965, 34, 143.
-
14Ashwell, G.; Annu. Rev. Biochem 1964, 33, 101.
-
15Dutcher, J. D.; Adv. Carbohydr. Chem. 1963, 18, 259.
-
16Pfrengle, F.; Reissig, H-U.; Chem. Soc. Rev 2010, 39, 549.
-
17Kirschning, A.; Jesberger, M.; Schoning, K.; Synthesis 2001, 4, 507.
-
18Xie, J; Carbohydrate Res. 2003, 338, 399.
-
19Levites-Agababa, E.; Menhaji, E.; Perlson, L. N.; Rojas, C. M.; Org. Lett. 2002, 4, 863.
-
20Chen, N.; Xie, J.; Molecules 2018, 23, 641.
-
21Davidson, M. H.; McDonald, F. E.; Org. Lett. 2004, 6, 1601.
-
22Velvadapu, V.; Andrade, R. B.; Carbohydr. Res 2008, 343, 145.
-
23Paixão, L.; Caldas, J.; Kloosterman, T. G.; Kuipers, O. P.; Vinga, S.; Neves, A. R.; Front Microbiol. 2015, 6, 1041.
-
24Kent, P. W.; Whitehouse, M. W.; Biochemistry of the aminosugars, Academic Press: New York, 1955.
-
25Ledderhose, G.; Zeitschr, F.; Physiol Chem 1878, 2, 213.
-
26Schmiedeberg, O.; Arch. F. Exp. Path. U. Pharm 1891, 28, 355.
-
27Moiler, F.; Z. Biol 1901, 42, 468.
-
28Levene, P. A.; LaForge, F. B.; J. Biol. Chem 1914, 18, 123.
-
29Gutiérrez-Moreno, N. J.; Medranob, F.; Yatsimirsky, A. K.; Org. Biomol. Chem 2012, 10, 6960.
-
30McGowan, J. V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J. M.; Yellon, D. M.; Cardiovasc. Drugs Ther 2017, 1, 63.
-
31Barata, L. E. S.; Cienc. Cult. (Sao Paulo) Supl. 1980, 32, 449.
-
32Lee, C.-H.; Schaffner, C.; Tetrahedron Lett 1966, 47, 5837.
-
33Els, M. J.; Ganem, B.; Carbohydr. Res 1988, 176, 316.
-
34Okubo, S.; Nakamura, N.; Morinoto, M.; Mineura, K.; Marumo, H.; Omura, S.; The Journal of Antibiotics 1980, 33, 221.
-
35Hamada, Y.; Shioiri, T.; Tetrahedron Lett. 1982, 23, 1193.
-
36Troy, F. A.; Encyclopedia Biol. Chem. 2004, 3, 407.
-
37Kleene, R.; Schachner, M.; Nat. Rev. Neurosci 2004, 5, 195.
-
38Dumitriu. S.; Polysaccharides: Structural Diversity and Functional,Versality: 2nd ed., Marcel Dekker: New York, 2005, Cap. 30.
-
39Bonfanti, L.; Prog. .Neurobiol. 2006, 80, 129.
-
40Gascon, E.; Vutskits, L.; Kiss, J. K.; Brain Res. Rev 2007, 56, 101.
-
41Miyata, S.; Sato, C.; Kitajima, K.; Trends Glycosci. Glyc. 2007, 19, 85.
-
42Janas, T.; Janas, T.; Biochim. Biophys. Acta 2011, 1808, 2923.
-
43Kimio, F.; Trends Glycosci. Glycotechnol. 2004, 89, 143.
-
44Huang, F.; Spiteller, D.; Koorbanally, N. A.; Li, Y.; Llewellyn, N. M.; Spencer, J. B.; ChemBioChem 2007, 8, 283.
-
45Ellis, G. P.; Honeyman, J.; J. Chem. Soc. 1952, 1490.
-
46Hermanson, G. T.; Bioconjugate Techniques 3rd ed., Academic Press: Cambridge 2013, cap. 2.
-
47Anderson, J. W.; Nicolosi, R. J.; Borzelleca, J. F.; Food Chem. Toxicol. 2005, 43, 187.
-
48Brautaset, T.; Sletta, H.; Nedal, A.; Borgos, S. E.; Degnes, K. F.; Bakke, I.; Volokhan, O.; Sekurova, O. N.; Treshalin, I. D.; Mirchink, E. P.; Dikiy, A.; Ellingsen, T. E.; Zotchev, S. B.; Chem Biol. 2008, 15, 1198.
-
49Muhizi, T.; Grelier, S.; Coma, V.; J. Agric. Food Chem. 2009, 57, 8770.
-
50Adam, R. D.; Clin. Microbiol. 2001, 14, 447.
-
51Milewski, S.; Gabriel, I.; Olchowy, J.; Yeast 2006, 23, 1.
-
52Kikuchi, K.; Tsuiki, S.; Biochim. Biophys. Acta. 1973, 327, 193.
-
53Chen, X.; Varki, A.; ACS Chem. Biol. 2010, 5, 163.
-
54Adak, A. K.; Yu, C. C.; Liang, C. F.; Lin, C. C.; Curr. Opin. Chem. Biol. 2013, 17, 1030.
-
55Vetter, N. D.; Langill, D. M.; Anjum, S.; Boisvert-Martel, J.; Jagdhane, R. C.; Omene, E.; Zheng, H.; van Straaten, K. E.; Asiamah, I.; Krol, E. S.; Sanders, D. A.; Palmer, D. R.; J. Am. Chem. Soc. 2013, 135, 5970.
-
56van Straaten, K. E.; Ko, J. B.; Jagdhane, R.; Anjum, S.; Palmer, D. R.; Sanders, D. A.; J. Biol. Chem 2013, 288, 34121.
-
57Iwai, Y.; Tanaka, H.; Oiwa, R.; Shimizu, S.; Omura, S.; Biochim. Biophys. Acta 1977, 498, 223.
-
58Janiak, A.; Milewski, S.; Med. Mycol. 2001, 39,401.
-
59Floss, H. G.; Yu, T. W.; Arakawa, K.; J. Antibiot. 2011, 64, 34.
-
60Arakawa, K.; Müller, R.; Mahmud, T.; Yu, T.W.; Floss, G.; J. Am. Chem. Soc. 2002, 124, 10644.
-
61Guo, J. T.; Frost, J. W.; J. Am. Chem. Soc. 2002, 124, 10642.
-
62Park, J. W.; Park, S. R.; Nepal, K. K.; Han, A. R.; Ban, Y. H.;Yoo, Y. J.; Kim, E. M.; Kim, D.; Sohng, J. K.; Yoon, Y. J.; Nat. Chem. Biol. 2011, 7, 843.
-
63Burgie, E. S.; Thoden, J. B.; Holden, H. M.; Protein Sci. 2007, 16, 887.
-
64Burgie, E. S.; Holden, H. M.; Biochemistry 2008, 47, 3982.
-
65Cook, P. D.; Holden, H. M.; Biochemistry 2008, 47, 2833.
-
66Nedal, A.; Sletta, H.; Brautaset, T.; Borgos, S. E. F.; Sekurova, O. N.; Ellingsen, T. E.; Zotchev, S. B.; Appl. Environ. Microbiol. 2007, 73, 7400.
-
67Otten, S. L.; Gallo, M. A.; Madduri, K.; Liu, X.; Hutchinson, C. R.; J. Bacteriol. 1997, 179, 4446.
-
68Hauser, F. M.; Ellenberger, S. R.; Chem. Rev. 1986, 86, 35.
-
69Guo, Z.; Li, J.; Qin, H.; Wang, M.; Lu, X.; Li, X.; Chen, Y.; Angew. Chem., Int. Ed. Engl. 2015, 54, 5175.
-
70Kumagai, A. H.; Yagita, A.; Akamatsu, N.; J. Antibiot. 1982, 34, 1571.
-
71Mirabella, S.; Cardona, F.; Goti, A.; Org. Biomol. Chem 2016, 14, 5186.
-
72Dahl, R. S.; Finney, N. S.; J. Am. Chem. Soc. 2004, 126, 8356.
-
73Buttar, S.; Caine, J.; Goné, E.; Harris, R.; Gillman , J.; Atienza, R.; Gupta , R. K. M.; Jain, L.; Abascal, N. C.; Levine, Y.; Repka, L. M.; Rojas, C. M.; J. Org. Chem., 2018, 83, 8054.
-
74Ansari, A. A.; Reddy, Y. S.; Vankar, Y. D.; Beilstein J. Org. Chem. 2014, 10, 300.
-
75Seeberger, P. H.; Roehrig, S.; Schell, P.; Wang, Y.; Christ, W. J.; Carbohydr. Res 2000, 328, 61.
-
76Liu, J.; Di Bussolo, V.; Gin, D. Y.; Tetrahedron Lett. 2003, 44, 4015.
-
77Schmidt, R. R.; Vankar, Y. D.; Acc. Chem. Res. 2008, 41, 1059.
-
78Rissé, S.; Roger, P.; Monneret, C.; J. Carbohydr. Chem. 1993, 12, 1105.
-
79Ferrier, R. J.; Collins, P. M.; Monosaccharides: Their chemistry and their roles in natural products Wiley & Sons: Chichester, 1995.
-
80Okazaki, H.; Hanaya, K.; Shoji, M.; Hada, N.; Sugai, T.; Tetrahedron 2013, 69, 7931.
-
81Poulain, F.; Serre, A.-L.; Lalot, J.; Leclerc, E.; Quirion, J.-C.; J. Org. Chem. 2008, 73, 2435.
-
82Poulain, F.; Leclerc, E.; Quirion, J.-C.; Tetrahedron Lett. 2009, 50, 1803.
-
83Okazaki, H.; Hanaya, K.; Shoji, M.; Hada, N.; Sugai, T.; Tetrahedron 2013, 69, 7931.
-
84Sobti, A. Sulikovski, G. A.; Tetrahedron Lett 1994, 35, 8259.
-
85Castilla, J.; Risquez, R.; Cruz, D.; Higaki, K.; Nanba, E.; Ohno, K.; Suzuki, Y.; Diaz, Y.; Mellet, C. O.; Garcia Fernandez, J. M.; Castillon, S.; J. Med. Chem 2012, 55, 6857.
-
86Ji, L.; Zhang, D.; Zhao, Q.; Hu, S.; Qian, C.; Chen, X.-Z.; Tetrahedron 2013, 69, 7031.
-
87Parker, K. A.; Chang, W.; Org. Lett. 2005, 7, 1785.
-
88Parker, K. A.; Chang, W.; Org. Lett. 2003, 5, 3891.
-
89Alcázar, E.; Pletcher, J. M.; McDonald, F. E.; Org. Lett. 2004, 6, 3877.
-
90Koo, B. S.; McDonald, F. E.; Org. Lett. 2007, 9, 1737.
-
91Davidson, M. H; McDonald, F. E.; Org. Lett. 2004, 6, 1601.
-
92Lafont, D.; Descotes, D.; Carbohydr. Res 1987, 166, 195.
-
93Lafont, D.; Guilloux, P.; Descotes, D.; Carbohydr. Res 1989, 61, 193.
-
94Lafont, D.; Woliny, A.; Boullanger, P.; Carbohydr. Res 1998, 310, 9.
-
95Mendlik, M. T; Tao, P.; Hadad, C. M.; Coleman, R. S.; Lowary, T. L.; J. Org. Chem. 2006, 71, 8059.
-
96Mirabella, S.; Cardona, F.; Goti, A.; Org. Lett. 2015, 17, 728.
-
97Ravindran, B.; Sakthivel, K.; Suresh, C. G.; Pathak, T.; J. Org. Chem. 2000, 65, 2637.
-
98Oliveira, R. N.; Cottier, L.; Sinou, D.; Srivastava, R. M.; Tetrahedron 2005, 61, 8271.
-
99Zhang, G.; Shi, L.; Liu, Q.; Wang, J.; Li, L.; Liu, X.; Tetrahedron 2007, 63, 9705.
-
100Baer, H. H.; Hanna, Z. S.; Can. J. Chem. 1981, 59, 889.
-
101Brito, T. M. B.; Silva, L. P.; Siqueira, V. L.; Srivastava, R. M.; J. Carbohydr. Chem. 1999, 18, 609.
-
102Mukherjee, A.; Jayaraman, N.; Carbohydr. Res 2013, 380, 51.
-
103Bussolo, V. D.; Romano. M. R.; Favero, L.; Pineschi, M.; Crott, P.; J. Org. Chem. 2006, 71, 1696.
-
104Liberek, B.; Dabrowska, A.; Frankowski, R.; Matuszewska, M.; Smiatacz, Z.; Carbohydr. Res 2002, 337, 1803.
-
105Ding, F.; William, R.; Wang, F.; Ma, J.; Ji, L.; Liu, X.-W.; Org. Lett. 2011, 13, 652.
-
106Ding, F.; William, R.; Wang, S.; Gorityala, B. K.; Liu, X.-W.; Org. Biomol. Chem 2011, 9, 3929.
-
107Yoshimura , J.; Adv. Carbohydr. Chem. Biochem. 1984, 42, 69.
-
108Grisebach, H.; Schmid, R.; Angew. Chem., Int. Ed. 1992, 11, 159.
-
109Hanessian, S.; Haskell, T. H. In The Carbohydrates; Pigman, W., Horton, D., eds.; Academic Press: New York, 1970, 139.
-
110Brimacombe, J. S.; Angew. Chem., Int. Ed. 1992, 8, 401.
-
111Holder, N. L.; Chem. Rev. 1982, 82, 287.
-
112Fraser-Reid, B.; Mclean, H.; Usherwood, E. W.; J. Am. Chem. Soc. 1969, 91, 5392.
-
113Fraser-Reid, B.; Mclean, H.; Usherwood, E. W.; Yunker, M.; Can. J. Chem. 1970, 48, 2877.
-
114Holder, N. L.; Fraser-Reid, B.; Can. J. Chem. 1973, 51, 3357 e referências nelas citadas.
-
115Fraser-Reid, B.; Walker, D. L.; Ten, S. Y-K.; Holder, N. L.; Can. J. Chem. 1973, 51, 3950.
-
116Srivastava, R. M.; Carthy, B. J.; Fraser-Reid, B.; Tetrahedron Lett. 1974, 2175.
-
117Apostolopoulos, C. D.; Couladouros, E. A.; Georgiadis, M. P.; Liebigs Ann Chem 1994, 781.
-
118Freitas Filho, J. R.; Srivastava, R. M.; Silva, W. J. P.; Cottier, L.; Sinou, D.; Carbohydr. Res 2003, 338, 673.
-
119Dahl, R. S.; Finney, N. S.; J. Am. Chem. Soc. 2004, 126, 8356.
-
120Dahl, R. S.; Baldridge, K. K.; Finney, N. S.; Synthesis 2010, 2292.
-
121Srivastava, R. M.; Souza, A. M. A.; Silva, L. P.; Freitas Filho, J. R.; Hallwass, F.; J. Braz. Chem. Soc. 2002, 13, 158.
-
122Freitas Filho, J. R.; Cottier, L.; Srivastava, R. M.; Sinou, D.; Synlett 2003, 1358.
-
123Ji, X-M.; Mo, J.; Liu, H-M.; Sun, H-P.; Carbohydr. Res 2006, 341, 2312.
-
124Zhu, Z.; Glazier, D. A.; Yang, D.; Tanga, W.; Adv. Synth. Catal 2018, 360, 2211.
-
125Ding, W.; Yu, J.-P.; Shi, X.-X.; Nie, L.-D.; Quan, N.; Li, F.-L. Tetrahedron: Asymmetry 2015, 26, 1037.
-
126Song, W.; Zhao, Y.; Lynch, J. C.; Kim, H.; Tang, W.; Chem. Commun. 2015, 51, 17475.
-
127Corsi, M.; Bonanni, M.;Catelani, G.; D’Andrea, F.; Gragnani, T.; Bianchini, R.; J. Org. Chem. 2013, 3, 41.
-
128Suami, T.; Machinami, T.; Hitsamatsu, H.; J. Med. Chem 1979, 22, 247.
-
129Omura, S.; Katagiri, M.; Atsumi, K.; Hata, T.; J. Chem. Soc Perkin 1, 1974, 0, 1627.
-
130Georges, M.; Mackay, D.; Fraser-Reid, B.; Can. J. Chem. 1984, 62, 1539.
-
131Futagami, S.; Ohashi, Y.; Imura, K.; Hosoya, T.; Ohmori, K.; Matsumoto, T.; Suzuki, K.; Tetrahedron Lett. 2000, 41, 1063.
-
132Brimbe, M. A.; Brenstrum, T, J.; Tetrahedron Lett 2000, 41, 1107.
-
133Johnson, A. W.; Smith, R. M.; Guthrie, R. D.; J. Chem. Soc. Perkin Trans 1 1972, 2153.
-
134Smith, G. R.; Giuliano, R. M.; Carbohydr. Res 2000, 323, 208.
-
135Willians, D. H.; Bardsley, B.; Angew. Chem., Int. Ed. 1999, 28, 1172.
-
136Fu, X.; Albermann, C.; Jiang, J.; Liao, J.; Zhang, C.; Thorson, J.; Nat. Biotechnology 2003, 21, 1467.
-
137Banoub, J.; Boullanger, P.; Lafont, D.; Chem. Rev. 1992, 92, 1167.
-
138Coben, M. L.; Science 1992, 257, 1050.
-
139Neu, H. C.; Science 1992, 257, 1064.
-
140Lin, H.; Walsh, C. T.; J. Am. Chem. Soc. 2004, 126, 13998.
-
141Chang, C.-W. T.; Hui, Y.; Elchert, B.; Wang, J.; Li, J.; Rai, R.; Org. Lett. 2002, 4, 4603.
-
142Elchert, B.; Li, J.; Wang, J.; Hui, Y.; Rai, R., Ptak, R.; Ward, P.; Takemoto, J. Y.; Bensaci, M.; Chang, C. -W. T.; J. Org. Chem. 2004, 69, 1513.
-
143O’Connor, S.; Lam, L. K. T.; Jones, N. D.; Chaney, M. O.; J. Org. Chem. 1976, 41, 2087.
-
144S uzuki, T.; Suzuki, S. T.; Yamada, I.; Koashi, Y.; Yamanda, K.; Chida, N.; J. Org. Chem. 2002, 67, 2874.
-
145McAuliffe, J. C.; Stick, R. V.; Stone, B. A.; Tetrahedron Lett 1996, 37, 2479.
-
146Amelung, W. In Assessment Methods for Soil Carbon; Lal, R., Kimble, J. M., Follett, R. F., Stewart, B. A., eds.; Lewis Publishers: Boca Raton, 2001, pp. 233-272.
-
147Hu , Y.; Zheng, Q.; Zhang, S.; Noll, L.; Wanek, W.; Soil Biol. Biochem. 2018, 123 , 115.
-
148Kandeler, E.; Tscherko, D.; Bruce, K. D.; Stemmer, M.; Hobbs, P. J.; Bardgett, R. D.; Amelung, W.; Biol. Fertil. Soils 2000, 32, 390.
-
149Brock, T. D.; Madigan, M. T.; Biology of Microorganisms, Prentice-Hall: Englewood Cliffs,1988.
-
150Cochran, T. W., Vercellotti, J. R.; Carbohydr. Res 1978, 61, 529.
-
151Chantigny, M. H.; Angers, D. A.; Prevost, D.; Vezina, L. P.; Chalifour, F. P.; Soil Sci. Soc. Am. J 1997, 61, 262.
-
152Zhanga, X.; Amelunga, W.; Yuanb, Y.; Samson-Liebigc, S.; Brown, L.; Zecha, W.; Appl. Soil Ecol. 1999, 11, 271.
-
153Solomon, D.; Lehmann, J.; Zech, W.; Biol. Fertil. Soils 2001, 33, 85.
-
154Turrión, M.-B.; Glaser, B.; Zech, W.; Biol. Fertil. Soils 2002, 35, 49.
-
155Amelung, W.; Zhang, X.; Flach, K.W.; Zech, W.; Soil Sci. Soc. Am. J 1999, 63, 86.
-
156Glaser, B.; Turrión, M-B.; Alef, K.; Soil Biol. Biochem. 2004, 36, 399.
-
157Yokoe, M.; Aoi, M.; Okada, M.; J. Polym. Sci., Part A: Polym. Chem 2005, 43, 3909.
-
158García-Martín, M. G.; Pérez, R. R; Hernández, E. B.; Espartero, J. L.; Muñoz-Guerra, S.; Galbis, J. A.; Macromolecules 2005, 38, 8664.
-
159Göpferich, A.; Biomaterials 1996, 17, 103.
-
160Cunliffe, D.; Pennadam, S.; Alexander, C.; Eur. Polym. J. 2004, 40, 5.
-
161Kiely, D. E.; Chen, L.; Lin, T. H.; J. Am. Chem. Soc. 1994, 116, 571.
-
162Kiely, D. E.; Chen, L.; Lin, T. H.; J. Polym. Sci., Part A: Polym. Chem 2000, 38, 594.
-
163Styron, S. D.; Kiely, D. E.; Ponder, G.; J. Carbohydr. Chem. 2003, 22, 123.
-
164Galbis, J. A.; García-Martín, M. G. In Monomers, polymers and composites from renewable resources; Belgacem, M. N., Gandini, A., eds.; Elsevier: Oxford., 2008, cap. 5.
-
165Galbis, J. A.; Martín, M. de G. G. M.; Paz M. V.; Galbis. E.; Chem. Rev. 2016, 116, 1600.
-
166Martín, M. G.; Perez, R. R.; Hernández, E. B.; Galbis, J. A.; Carbohydr. Res 2001, 333, 95.
-
167Jasinska, L.; Villani, M.; Wu, J.; Es, V. D.; Klop, E.; Rastogi, S.; Koning, C. E.; Macromolecules 2011, 44, 3458.
-
168Walc, L. J.; Villani, M.; Dudenko, D.; Asselen, O. V.; Klop, E.; Rastogi, S.; Hansen, M. R.; Koning, C. E.; Macromolecules 2012, 45, 2796.
-
169Gomez, R. V.; Varela, O.; Macromolecules 2009, 42, 8112.
-
170Lu, H.; Sun, P.; Zheng, Z.; Yao, X.; Wang, X.; Chang, F. C.; Polym. Degrad. Stab 2012, 97, 661.
-
171Bachmann, F.; Reimer, J.; Ruppenstein, M.; Thiem, J.; Macromol. Chem. Phys 2001, 202, 3410.
Datas de Publicação
-
Publicação nesta coleção
15 Ago 2019 -
Data do Fascículo
Jun 2019
Histórico
-
Recebido
17 Dez 2018 -
Aceito
14 Mar 2019 -
Publicado
17 Abr 2019