Resumo
Pantoprazole is a proton pump inhibitor used in the treatment of digestive ulcers, gastro-esophageal reflux disease and in the eradication of Helicobacter pylori. In this work, an analytical method was developed and validated for the quantification of sodium pantoprazole by HPLC. The method was specific, linear, precise and exact. In order to verify the stability of pantoprazole during dissolution assays, pantoprazole solution in phosphate buffer pH 7.4 was kept at room temperature and protected from light for 22 days. Pantoprazole presented less than 5% of degradation in 6 hours and the half live of the degradation was 124 h.
pantoprazole; validation; stability
pantoprazole; validation; stability
NOTA TÉCNICA
Validação de metodologia analítica por cromatografia líquida para doseamento e estudo da estabilidade de pantoprazol sódico
Validation of analytical methodology by hplc for quantification and stability evaluation of sodium pantoprazole
Renata Platcheck RaffinI; Letícia Marques ColoméI; Sílvia Stanisçuaski GuterresI,* * e-mail: nanoc@farmacia.ufrgs.br ; Adriana Raffin PohlmannII
IFaculdade de Ciências Farmacêuticas, Universidade Federal do Rio Grande do Sul, Av. Ipiranga, 2752, 90610-000 Porto Alegre RS, Brasil
IIDepartamento de Química Orgânica, Instituto de Química, Universidade Federal do Rio Grande do Sul, CP 15003, 91501-970 Porto Alegre - RS, Brasil.
ABSTRACT
Pantoprazole is a proton pump inhibitor used in the treatment of digestive ulcers, gastro-esophageal reflux disease and in the eradication of Helicobacter pylori. In this work, an analytical method was developed and validated for the quantification of sodium pantoprazole by HPLC. The method was specific, linear, precise and exact. In order to verify the stability of pantoprazole during dissolution assays, pantoprazole solution in phosphate buffer pH 7.4 was kept at room temperature and protected from light for 22 days. Pantoprazole presented less than 5% of degradation in 6 hours and the half live of the degradation was 124 h.
Keywords: pantoprazole; validation; stability.
INTRODUÇÃO
O pantoprazol [5-(difluorometoxi)-2-[[(3,4-dimetoxi-2-piridinil)metil]sulfinil]-1H-benzimidazol] é um fármaco inibidor da bomba de prótons, atualmente considerado um dos mais importantes para tratamento de uma variedade de doenças relacionadas ao trato gastrintestinal superior1. Este fármaco vem sendo amplamente utilizado no tratamento de úlceras gástricas, refluxo gastro-esofágico e condições hipersecretórias patológicas, devido à sua capacidade de inibir irreversivelmente a bomba de prótons, diminuindo a secreção gástrica proveniente das células parietais do estômago2. Outra indicação é o tratamento adjunto a antibióticos para erradicação de Helicobacter pylori3.
O mecanismo de ação do pantoprazol, um pró-fármaco, baseia-se na sua ativação no ambiente ácido das células parietais gástricas, após sua absorção intestinal e circulação na corrente sangüínea. A ativação leva à formação de uma sulfonamida catiônica cíclica (Figura 1), que se liga a uma ou mais subunidades catalíticas da bomba de prótons, alterando sua configuração e provocando uma inibição irreversível do transporte ácido. No entanto, para que o pantoprazol esteja apto para atuar em seu local de ação, a ativação deve ocorrer somente nos canalículos das células parietais, sendo necessária sua passagem intacta pelo lúmen do estômago2.
O sal sódico sesquiidratado é a forma mais utilizada do fármaco (C16H14F2N3 O4SNa.1,5H2O), sendo solúvel em água, pouco solúvel em tampão fosfato pH 7,4 e insolúvel em n-hexano1. A degradação do pantoprazol em soluções aquosas é dependente do pH, sendo que a taxa de degradação aumenta com o abaixamento do pH. Jungnickel1 relatou que, em pH 5, a meia vida de degradação é de aproximadamente 2,8 h e em pH 7,8 é de 220 h.
O pantoprazol também apresenta instabilidade frente à presença de diversos sais em solução aquosa, como cloreto de sódio, ácido cítrico, citrato trissódico, citrato monossódico, bicarbonato de sódio e carbonato de cálcio, em diferentes concentrações e valores de pH, conforme descreveram Ekpe e Jacobsen4. Os autores observaram ainda que as condições de armazenamento mais adequadas para diminuir a degradação do fármaco são pH 10 e 4 ºC.
Em outro estudo, realizado por Detinger e colaboradores5, comprimidos foram triturados e o pó suspenso em solução aquosa de bicarbonato (2 mg/mL de pantoprazol) com o objetivo de preparar uma forma farmacêutica líquida contendo pantoprazol. A suspensão extemporânea foi avaliada quanto à sua estabilidade por 62 dias sob refrigeração (2 a 8 ºC). Após este período, mais de 90% da quantidade inicial de pantoprazol manteve-se estável. A mesma metodologia de preparação da solução extemporânea foi estudada em escala maior por Ferron e colaboradores6. Neste estudo, 34 comprimidos foram triturados e suspensos em 680 mL de solução de bicarbonato a 4,2% e mantidos em seringas plásticas. A 25 ºC a suspensão foi estável por 1 dia (99,1%), a 5 ºC por 2 semanas (98,6%) e a 20 ºC por até 3 meses de armazenamento (98,3%).
Em outro trabalho7, o pó liofilizado comercial de pantoprazol foi reconstituído com solução de NaCl a 0,9% com o objetivo de estudar sua estabilidade, visando o uso por via intravenosa. A solução (4 mg/mL) foi mantida em seringas de polipropileno à temperatura ambiente por 96 h. A concentração final de pantoprazol em solução foi 96% da concentração inicial, porém a solução alterou sua cor para laranja-amarelado. O autor considerou que o grau de degradação não foi significativo, não havendo alteração da concentração do fármaco nas condições testadas.
De forma geral, os estudos mostram que a degradação do pantoprazol aumenta com a presença de sais em solução e com a diminuição do pH, o que reafirma a necessidade de administrá-lo em uma forma gastrorresistente, capaz de permitir absorção exclusivamente entérica. Normalmente, o planejamento de novas formas farmacêuticas deve contemplar como etapa inicial o desenvolvimento e validação de metodologia analítica capaz de quantificar o fármaco contido nas formulações, bem como eventuais produtos de degradação formados, especialmente quando essas metodologias estão ausentes em códigos oficiais. Apesar do pantoprazol já ser um fármaco bastante usado na terapêutica e ter sido aprovado pelo FDA em 2000, sua monografia ainda não está descrita em compêndio oficial algum.
Para o controle de qualidade de formas farmacêuticas, os testes de dissolução preconizados pelos códigos oficiais são indispensáveis, sendo importantes também para orientar o desenvolvimento de novas formulações. Além disso, permitem a demonstração in vitro de características da formulação, as quais podem estar correlacionadas com seu desempenho in vivo. Em geral, estes testes são conduzidos com velocidade e agitação padronizadas e sob condições "sink", mimetizando, tanto quanto possível, as condições do trato gastrintestinal8. Estes podem ser conduzidos em aparatos, como dissolutores9,10, nos quais as formas farmacêuticas são colocadas em recipientes com volume padronizado de meio de dissolução e mantidas até sua completa dissolução ou em equipamentos de células de fluxo9, onde a amostra é submetida a um fluxo de meio de dissolução até seu esgotamento. Quando se utiliza qualquer uma destas metodologias, o baixo limite de quantificação do método analítico empregado no doseamento do fármaco é fundamental para garantir precisão na avaliação dos pontos iniciais e finais da dissolução.
Os ensaios de dissolução para novas formas farmacêuticas de liberação prolongada devem ser realizados em meio capaz de mimetizar as condições intestinais. Desta forma, os ensaios podem ser realizados empregando-se soluções tamponadas (pH 7,4) por, no mínimo, 2 h. A estabilidade da substância no meio deve ser garantida, especialmente no caso das formas farmacêuticas que utilizam polímeros de liberação colônica, cuja solubilidade se dá em solução com pH superior a 78,10. No caso de formulações revestidas e/ou gastrorresistentes contendo pantoprazol, o estudo da sua estabilidade no meio de dissolução é fundamental para se detectar se há ou não degradação do pantoprazol após sua liberação/dissolução, o que ocasionaria interpretação errônea do perfil de liberação.
Para a quantificação do pantoprazol em estudos de dissolução e estabilidade, o limite de quantificação é o parâmetro mais importante na escolha do método. Entre os métodos já existentes na literatura para determinação do pantoprazol em formas farmacêuticas, um método5 de cromatografia líquida de alta eficiência desenvolvido para determinação da estabilidade de pantoprazol em suspensão oral extemporânea utilizou fase móvel em sistema isocrático, constituída de acetonitrila e fosfato de potássio dibásico 50 mM, com faixa de análise entre 6 e 24 µg/mL. Outro método cromatográfico semelhante11 para determinação de pantoprazol em comprimidos utilizou fase móvel constituída de acetonitrila (300 mL) e tampão fosfato pH 7,4 (600 mL). Os autores utilizaram coluna octadecilsilano (C18) e trabalharam na faixa de análise entre 5 e 20 µg/mL. Em ambos os métodos, o limite de quantificação não foi determinado.
Um método que empregou coluna octilsilano (C8) e fase móvel isocrática constituída de 70:30 tampão e acetonitrila, sendo o tampão constituído por 0,02 M de fosfato de sódio monobásico e 0,03 M de fosfato de sódio dibásico, foi validado. No entanto, os autores descreveram uma concentração inferior de trabalho alta para o pantoprazol (228 µg/mL)12. A especificidade foi avaliada injetando-se uma mistura dos excipientes dos comprimidos comerciais de pantoprazol.
Considerando o exposto, este trabalho teve como objetivo desenvolver e validar uma metodologia analítica para doseamento de pantoprazol, através de cromatografia líquida de alta eficiência, a fim de obter um limite de quantificação, que permita a aplicação do método em estudos de dissolução in vitro. O trabalho visou também aplicar esta metodologia analítica na quantificação do pantoprazol em breve estudo de estabilidade do fármaco em tampão fosfato pH 7,4, a fim de investigar a presença de produtos de degradação que possam ser formados durante os ensaios de dissolução.
PARTE EXPERIMENTAL
Materiais e equipamentos
Pantoprazol sódico sesquiidratado (99,84%, Henrifarma, São Paulo, Brasil); acetonitrila grau CLAE; água purificada através de sistema Milli-Q para uso em CLAE; fosfato de potássio monobásico; fosfato de potássio dibásico. Cromatógrafo líquido de alta eficiência Perkin Elmer Série 200 acoplado a detector UV-VIS. Cromatógrafo líquido de alta eficiência Schimadzu LC-10ADV acoplado a detector de arranjo de diodo (SPD-M10AVP).
Metodologia
A validação da metodologia analítica foi realizada segundo os critérios propostos pela ICH ("International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use")13 de 1996, USP10 e resolução da Anvisa RE nº 899, de 29/5/200314. Os parâmetros avaliados foram: linearidade, precisão intermediária, repetibilidade, exatidão, limites de detecção e quantificação e especificidade frente aos produtos de degradação, sendo os últimos ensaios realizados em cromatógrafo Perkin Elmer Série 200 com detector UV/VIS. A pureza do pico foi avaliada em cromatógrafo acoplado a detector de arranjo de diodo.
Os parâmetros cromatográficos utilizados em ambos os equipamentos encontram-se na Tabela 1.
O tampão fosfato pH 7,4, utilizado na fase móvel, foi preparado com 839 mg de fosfato de potássio monobásico e 137 mg de fosfato de potássio dibásico para 1000 mL de solução.
Para avaliação da linearidade, a faixa de concentração de pantoprazol utilizada nas curvas foi de 0,5 a 20,0 µg/mL, sendo a última diluição realizada em fase móvel. As amostras foram filtradas através de membrana com 0,22 µm de poro (Durapore, Millipore®) antes das análises e injetadas em triplicata no cromatógrafo, registrando-se os valores das áreas. Os limites de detecção e quantificação (Equações 1 e 2) foram calculados matematicamente através da relação entre o desvio padrão da curva de calibração e sua inclinação, usando o fator multiplicador sugerido pela norma da ICH13.

onde LD é o limite de detecção, LQ o limite de quantificação, DP o desvio padrão da reta de calibração e B o coeficiente angular da reta de calibração.
Para avaliar a repetibilidade, três concentrações diferentes (6, 9 e 14 µg/mL) foram injetadas em triplicata, totalizando nove determinações, sendo o resultado da análise expresso em termos de desvio padrão relativo (DPR). A precisão intermediária foi avaliada através da comparação entre as injeções de uma mesma concentração (9 µg/mL) realizadas em três dias consecutivos, calculando-se posteriormente o DPR dos resultados das determinações.
O teste de recuperação (exatidão) foi realizado adicionando-se uma quantidade conhecida de fármaco (2 µg/mL) à solução-amostra, resultando nas concentrações de 6, 9 e 14 µg/mL. As soluções-amostra utilizadas para avaliações da precisão, da repetibilidade e da exatidão foram preparadas a partir de micropartículas contendo pantoprazol. Tais partículas foram preparadas pelo método de evaporação do solvente, após a emulsificação de uma solução de acetona contendo pantoprazol e Eudragit S100® com óleo mineral contendo monooleato de sorbitano, na proporção 1:1 (m/m) polímero:fármaco. Depois de formadas, as micropartículas foram recolhidas por filtração e lavadas com cicloexano.
Para avaliação da estabilidade e verificação da especificidade do método frente a produtos de degradação, o pantoprazol foi exatamente pesado (25 mg), dissolvido em água e o volume completado a 25 mL. Uma nova diluição foi realizada em tampão fosfato pH 7,4 obtendo-se uma concentração de 50 µg/mL4. As amostras, mantidas ao abrigo da luz e do calor, foram analisadas por CLAE em um período de 1 h até 22 dias, sendo que para análise, 1 mL da solução amostra foi dissolvido a 5 mL em fase móvel e filtrada para posterior injeção no cromatógrafo.
O perfil de dissolução das micropartículas foi realizado em equipamento dotado de célula de fluxo com mecanismo para movimento pendular, acoplado à bomba peristáltica, em banho termostatizado a 37 ºC. O meio de dissolução utilizado foi tampão fosfato pH 7,4 com fluxo de 1 mL/min. As coletas foram realizadas até as alíquotas estarem abaixo do limite de quantificação do método analítico.
RESULTADOS E DISCUSSÃO
A fase móvel composta de acetonitrila e tampão fosfato pH 7,4 na proporção 35:65 (v/v) foi considerada adequada. O pico referente ao pantoprazol (Figura 2) apresentou-se bem definido e com tempo de análise reduzido (8 min) em relação ao descrito na literatura para método empregando gradiente de solventes na fase móvel (28 min)15. O cromatograma obtido da amostra após 168 h em tampão fosfato foi utilizado para verificar a seletividade do método analítico e pode-se verificar a completa separação dos picos do fármaco e dos produtos de degradação (Figura 2).
O estudo da linearidade do método analítico foi realizado através de análise de variância, verificando-se regressão linear significativa e desvio da linearidade não significativo (p<0,01) com injeção de concentrações compreendidas entre 0,5 e 20 µg/mL, em triplicata. O coeficiente de correlação encontrado para a curva média foi de 0,9994 (Tabela 2).
Com os dados provenientes da avaliação da linearidade foram calculados os limites de detecção e quantificação através das Equações 1 e 2, respectivamente. Os valores obtidos foram 0,182 µg/mL para o limite de detecção e 0,553 µg/mL para o limite de quantificação. Esse limite de quantificação permite a análise do fármaco até praticamente sua completa dissolução. Alguns métodos15,16 analíticos utilizando CLAE para determinação de pantoprazol em plasma obtiveram limites de detecção inferiores (0,05 e 0,025 µg/mL), porém utilizavam gradiente de fase móvel. Métodos que utilizam eluição em gradiente são demorados por necessitarem de longos períodos para equilibrar a coluna, maior gasto de solvente e bombas especializadas17.
Comparando-se as faixas de análise e limites de quantificação encontrados na literatura com os utilizados e determinados em nosso estudo, a metodologia aqui descrita reduz em 93% a faixa de análise, em relação à descrita por Badwan e colaboradores12 que passa de 228 a 670 µg/mL para 0,5 a 20 µg/mL. Essa redução torna o método viável para sua aplicação, tanto na quantificação da liberação do fármaco a partir de formas farmacêuticas quanto em seus estudos de estabilidade.
Os resultados de repetibilidade e precisão intermediária foram expressos em termos de desvio padrão relativo (DPR) (Tabela 3). A repetibilidade apresentou DPR de 1,71; 0,59 e 1,10 para as triplicatas das concentrações de 6, 9 e 14 µg/mL, respectivamente. A precisão intermediária foi avaliada em três dias consecutivos, com amostras de mesma concentração (9 µg/mL). Os valores de DPR foram 0,59; 0,54 e 0,27 para cada dia. Para ambos os estudos os valores de desvios padrões relativos foram inferiores a 2, demonstrando repetibilidade e precisão intermediária adequadas para o método analítico em questão. No que diz respeito à exatidão, o método permitiu a recuperação de 95,39 ± 3,77% para a concentração final de 6 µg/mL; 101,13 ± 1,71% para 9 µg/mL e 101,38 ± 1,46% para 14 µg/mL, o que caracteriza o método como exato, segundo os preceitos da "International Conference on Harmonization"13.
A análise dos cromatogramas demonstrou que há completa separação entre os picos do fármaco (6,7 min) e seus produtos de degradação em tampão fosfato (1,6 e 3,2 min), não havendo interferência dos produtos de degradação na quantificação do pantoprazol (Figura 2). A análise do pico referente ao fármaco, utilizando o detector de arranjo de diodo, demonstrou 100% de pureza no pico.
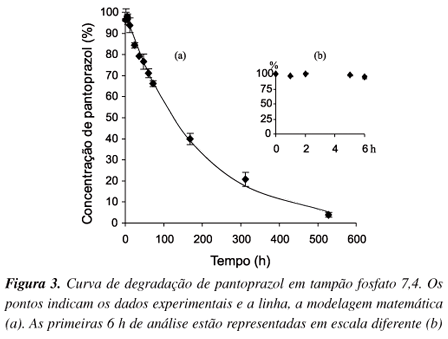
Através do estudo de estabilidade, verificou-se que a concentração de pantoprazol atinge 90% da concentração inicial em cerca de 19 h, em tampão fosfato pH 7,4, sendo que após 22 dias, a concentração atinge apenas 3,9% da concentração inicial (Figura 3). Por outro lado, o pantoprazol mantém 100% de sua concentração até 6 h, tempo normalmente limite para testes de dissolução10, pois existe a necessidade de mimetizar condições fisiológicas de trânsito no trato gastrintestinal.
A modelagem matemática da curva de degradação foi realizada empregando o software Micromath Scientist® segundo o modelo monoexponencial (Equação 3).

O valor da constante observada k foi 0,00558 ± 0,00015 h-1. O coeficiente de correlação foi de 0,9997. O tempo de meia-vida de degradação do pantoprazol em tampão fosfato foi de 124 h.
Com o objetivo de demonstrar a adequabilidade do método analítico, as micropartículas de pantoprazol empregadas para avaliação de precisão, repetibilidade e exatidão foram avaliadas quanto ao perfil de dissolução in vitro do pantoprazol em equipamento de células de fluxo, utilizando como meio tampão fosfato pH 7,4. O perfil de liberação do pantoprazol a partir das micropartículas foi determinado por 120 min (Figura 4), tendo demonstrado que neste período a totalidade de pantoprazol encapsulado foi liberada a partir das micropartículas.
CONCLUSÃO
A metodologia analítica proposta para detecção e quantificação de pantoprazol por CLAE mostrou-se sensível, precisa e linear na faixa de concentração entre 0,5 e 20 µg/mL, sendo adequada para avaliação do fármaco em formas farmacêuticas como micropartículas e em soluções de tampão fosfato. Além disso, esse limite de quantificação (0,553 µg/mL) torna a metodologia adequada à utilização em ensaios de dissolução e estabilidade. A estabilidade do fármaco em tampão fosfato pH 7,4 é limitada, porém suficiente para a realização de análises normalmente conduzidas em estudos biofarmacêuticos.
AGRADECIMENTOS
CNPq/MCT, CAPES e FAPERGS.
Recebido em 10/3/06; aceito em 4/8/06; publicado na web em 26/3/07
- 1. Jungnickel, P. W.; Clin. Ther 2000, 22, 1268.
- 2. Cheer, S.; Prakash, A.; Faulds, D.; Lamb, H.; Drugs 2003, 63, 101.
- 3. Poole, P.; Am. J. Health Syst. Pharm. 2001, 58, 999.
- 4. Ekpe, A.; Jacobsen, T.; Drug. Dev. Ind. Pharm. 1999, 25, 1057.
- 5. Detinger, P.; Swewnson, C.; Anaizi, N.; Am. J. Health Syst. Pharm. 2002, 59, 953.
- 6. Ferron, G.; Ku, S.; Abell, M.; Unruh, M.; Getsy, J.; Mayer, P.; Paul, J.; Am. J. Health Syst. Pharm 2003, 60, 1324.
- 7. Johnson, C.; Am. J. Health Syst. Pharm 2005, 62, 2410.
- 8. Aulton, M. E.; Delineamento de formas farmacêuticas, Artmed: Porto Alegre, 2005.
- 9. British Pharmacopoeia; Her Majesty's Stationery Office: Londres, 2000.
-
10Pharmacopoeia of the United States of America; 28th ed., Rockville Pharmacopoeial Convention: Estados Unidos da América, 2004.
- 11. Mansour, A. M.; Sorour, O. M.; Chromatographia 2001, 53 suppl., S478.
- 12. Badwan, A. A.; Nabulsi, L. N.; Omari, A. L.; Daraghmeh, M. M.; Ashour, M.; Abdou, A. M.; Jaber, A. M. Em Analytical Profiles of Drug Substances and Excipients; Florey, K.; Brittain, H.G., eds.; Academic Press, Elsevier Science: USA, 2002, vol. 29, p. 213-259.
-
13Guidance for Industry Q2B Validation of Analytical Procedures: Methodology; European Agency for the Evaluation of Medicinal Products (ICH): Londres, 1996.
-
14Agência Nacional de Vigilância Sanitária, resolução RE nº 899 de 23 de maio de 2003, Brasil.
- 15. Huber, R.; Müller, W.; J. Chromatogr 1990, 529, 389.
- 16. Xie, Z.; Chen, X.; Jin, F.; Zhong, D.; J. Chromatogr. Sci 2005, 43, 271.
- 17. Hutabarat, L. S.; Mulholland, M.; Greenfield, H.; J. Chromatogr., A 1998, 795, 377.
Datas de Publicação
-
Publicação nesta coleção
10 Ago 2007 -
Data do Fascículo
Ago 2007
Histórico
-
Aceito
04 Ago 2006 -
Recebido
10 Mar 2006