Resumos
O advento de medicamentos antidepressivos tornou a depressão um problema médico, passível de tratamento. Nas últimas cinco décadas, a psicofarmacologia da depressão evoluiu muito e rapidamente. Os primeiros antidepressivos - os antidepressivos tricíclicos (ADTs) e os inibidores da monaminooxidase (IMAOs) - foram descobertos através da observação clínica. Os ADTs apresentavam boa eficácia devido à sua ação, aumentando a disponibilidade de norepinefrina e serotonina. Seu uso foi limitado em função do bloqueio de receptores de histamina, colinérgicos e alfa-adrenérgicos que acarretavam efeitos colaterais levando à baixa tolerabilidade e risco de toxicidade. Da mesma forma, o uso dos IMAOs ficava comprometido em função do risco da interação com tiramina e o risco de crises hipertensivas potencialmente fatais. A nova geração de antidepressivos é constituída por medicamentos que agem em um único neurotransmissor (como os inibidores seletivos de recaptação de serotonina ou de noradrenalina) ou em múltiplos neurotransmissores/receptores, como venlafaxina, bupropion, trazodona, nefazodona e mirtazapina, sem ter como alvo outros sítios receptores cerebrais não relacionados com a depressão (tais como histamina e acetilcolina). Este artigo revisa a farmacologia dos antidepressivos, particularmente quanto ao mecanismo de ação, farmacocinética, efeitos colaterais e interações farmacológicas.
Psicofarmacologia; antidepressivos; mecanismo de ação; efeitos colaterais; farmacocinética; interação farmacológica
Antidepressant drugs turned depression into a treatable medical problem. In the last five decades, the psychopharmacology of depression has evolved rapidly. Early antidepressants - tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs) - were discovered through clinical observation. The TCAs exhibited good antidepressant efficacy due to the enhancement in serotonin and norepinephrine availability. Its use was limited because of unwanted side effects and toxicity risk related to the blockade of histaminergic, cholinergic and alfa-adrenergic receptors. MAOIs can interact with tyramine to cause potentially lethal hypertension and present potentially dangerous interactions with various medications and over-the-counter drugs. The new generation of antidepressants includes the single-receptor selective serotonin or norepinephrine inhibitors and the multiple-receptor-acting antidepressants, such as venlafaxine, bupropion, trazodone, nefazodone, and mirtazapine. They do not act on other receptor sites not related to depression (such as histamine or acetilcholine). This paper reviews the pharmacology of antidepressants, including its mechanism of action, pharmacokinetics, side effects and drug-drug interactions.
Psychopharmacology; antidepressants; mechanism of action; side effects; pharmacokinetics; drug interactions
Psicofarmacologia de antidepressivos
Ricardo Alberto Moreno1 1 . Coordenador do Grupo de Doenças Afetivas e Chefe da Coordenadoria de Atendimentos Especializados do Instituto de Psiquiatria do Hospital das Clínicas da FMUSP. , Doris Hupfeld Moreno2 1 . Coordenador do Grupo de Doenças Afetivas e Chefe da Coordenadoria de Atendimentos Especializados do Instituto de Psiquiatria do Hospital das Clínicas da FMUSP. e
Márcia Britto de Macedo Soares3 1 . Coordenador do Grupo de Doenças Afetivas e Chefe da Coordenadoria de Atendimentos Especializados do Instituto de Psiquiatria do Hospital das Clínicas da FMUSP.
RESUMO
O advento de medicamentos antidepressivos tornou a depressão um problema médico, passível de tratamento. Nas últimas cinco décadas, a psicofarmacologia da depressão evoluiu muito e rapidamente. Os primeiros antidepressivos os antidepressivos tricíclicos (ADTs) e os inibidores da monaminooxidase (IMAOs) foram descobertos através da observação clínica. Os ADTs apresentavam boa eficácia devido à sua ação, aumentando a disponibilidade de norepinefrina e serotonina. Seu uso foi limitado em função do bloqueio de receptores de histamina, colinérgicos e alfa-adrenérgicos que acarretavam efeitos colaterais levando à baixa tolerabilidade e risco de toxicidade. Da mesma forma, o uso dos IMAOs ficava comprometido em função do risco da interação com tiramina e o risco de crises hipertensivas potencialmente fatais. A nova geração de antidepressivos é constituída por medicamentos que agem em um único neurotransmissor (como os inibidores seletivos de recaptação de serotonina ou de noradrenalina) ou em múltiplos neurotransmissores/receptores, como venlafaxina, bupropion, trazodona, nefazodona e mirtazapina, sem ter como alvo outros sítios receptores cerebrais não relacionados com a depressão (tais como histamina e acetilcolina). Este artigo revisa a farmacologia dos antidepressivos, particularmente quanto ao mecanismo de ação, farmacocinética, efeitos colaterais e interações farmacológicas.
DESCRITORES
Psicofarmacologia; antidepressivos; mecanismo de ação; efeitos colaterais; farmacocinética; interação farmacológica
ABSTRACT
Antidepressant drugs turned depression into a treatable medical problem. In the last five decades, the psychopharmacology of depression has evolved rapidly. Early antidepressants tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs) were discovered through clinical observation. The TCAs exhibited good antidepressant efficacy due to the enhancement in serotonin and norepinephrine availability. Its use was limited because of unwanted side effects and toxicity risk related to the blockade of histaminergic, cholinergic and alfa-adrenergic receptors. MAOIs can interact with tyramine to cause potentially lethal hypertension and present potentially dangerous interactions with various medications and over-the-counter drugs. The new generation of antidepressants includes the single-receptor selective serotonin or norepinephrine inhibitors and the multiple-receptor-acting antidepressants, such as venlafaxine, bupropion, trazodone, nefazodone, and mirtazapine. They do not act on other receptor sites not related to depression (such as histamine or acetilcholine). This paper reviews the pharmacology of antidepressants, including its mechanism of action, pharmacokinetics, side effects and drug-drug interactions.
KEYWORDS
Psychopharmacology; antidepressants; mechanism of action; side effects; pharmacokinetics; drug interactions
Introdução
A descoberta no final da década de 50 de drogas antidepressivas e sua utilização na prática clínica trouxe um avanço importante no tratamento e no entendimento de possíveis mecanismos subjacentes aos transtornos depressivos.1,2 Tornou a depressão um problema médico passível de tratamento, semelhante a outras doenças como o diabetes e a hipertensão arterial. Até os anos 80 havia duas classes de antidepressivos, os tricíclicos (ADTs) e os inibidores de monoaminooxidase (IMAOs). Embora muito eficazes, apresentavam efeitos colaterais indesejáveis causados pela inespecificidade de sua ação farmacológica e eram potencialmente letais em casos de superdosagem.3 Nas últimas duas décadas surgiram novas classes de antidepressivos a partir da pesquisa de moléculas desprovidas dos efeitos colaterais dos heterocíclicos. Eles diferem dos clássicos ADTs e IMAOs, irreversíveis pela seletividade farmacológica, modificando e atenuando os efeitos colaterais.2
Os antidepressivos não influenciam de forma acentuada o organismo normal em seu estado basal, apenas corrigem condições anômalas. Em indivíduos normais não provocam efeitos estimulantes ou euforizantes como as anfetaminas. Aproximadamente 70% dos pacientes com depressão se beneficiam com os ADTs, mas 30% a 40% falham na resposta ao primeiro ensaio farmacológico, necessitando outra classe de antidepressivos ou mesmo eletroconvulsoterapia.4
Apesar dos avanços na pesquisa não dispomos de uma explicação completa e adequada do funcionamento dos antidepressivos, e assim servimo-nos de hipóteses para entender seu mecanismo de ação. Antidepressivos com estruturas químicas diferentes possuem em comum a capacidade de aumentar agudamente a disponibilidade sináptica de um ou mais neurotransmissores, através da ação em diversos receptores e enzimas específicos. Apesar de essencial, este efeito não explica a demora para se obter resposta clínica (de 2 a 4 semanas em média), sugerindo que a resolução dos sintomas da depressão requeira mudanças adaptativas na neurotransmissão.2 A principal teoria aceita para explicar tal demora é a da subsensibilização dos receptores pós-sinápticos. O aumento dos níveis de neurotransmissores por inibição da MAO ou bloqueio das bombas de recaptura de monoaminas resulta nesta subsensibilização, cuja resolução se correlaciona com o início da melhora clínica.2
Serão discutidos aspectos farmacológicos dos antidepressivos disponíveis no Brasil, mecanismos de ação propostos, farmacocinética, perfil de efeitos colaterais e interações farmacológicas.
Classificação
Os antidepressivos podem ser classificados de acordo com a estrutura química ou as propriedades farmacológicas. A estrutura cíclica (anéis benzênicos) caracteriza os antidepressivos heterocíclicos (tricíclicos e tetracíclicos). Os ADTs se dividem em dois grandes grupos: as aminas terciárias (imipramina, amitriptilina, trimipramina e doxepina) e as aminas secundárias (desmetilimipramina, nortriptilina e protriptilina). Maprotilina e amoxapina são antidepressivos tetracíclicos. As características farmacológicas da maprotilina se assemelham aos ADTs e ela será abordada dentro desta classe de antidepressivos.
Atualmente os antidepressivos, preferencialmente, são classificados em função da ação farmacológica, mais útil na prática clínica porque os antidepressivos de nova geração não compartilham estruturas comuns. Atualmente podemos dividi-los de acordo com o mecanismo de ação proposto, aumentando a eficiência sináptica da transmissão monoaminérgica (particularmente de neurônios noradrenégicos e/ou serotonérgicos). Medicamentos antidepressivos produzem aumento na concentração de neurotransmissores na fenda sináptica através da inibição do metabolismo, bloqueio de recaptura neuronal ou atuação em autoreceptores pré-sinápticos5 (tabela 1).
Antidepressivos Inibidores da Monoaminooxidase (IMAOs)
Mecanismo de ação
O mecanismo de ação dos IMAOs foi pouco estudado e ainda não está totalmente esclarecido.6 Sabe-se que a atividade da enzima monoaminoxidase (MAO) está inibida. Os subtipos da MAO, A e B, estão envolvidos no metabolismo de serotonina, noradrenalina e dopamina. Isocarboxazida, fenelzina e tranilcipromina são IMAOs não seletivos que se ligam de forma irreversível às MAOs A e B (tabela 1). A redução na atividade da MAO resulta em aumento na concentração desses neurotransmissores nos locais de armazenamento no sistema nervoso central (SNC) e no sistema nervoso simpático. O incremento na disponibilidade de um ou mais neurotransmissores tem sido relacionado à ação antidepressiva dos IMAOs. A inibição não seletiva dos IMAOs fenelzina, isocarboxazida e tranilcipromina resulta em subsensibilização de receptores a2- ou b-adrenérgicos e de serotonina. Possivelmente as mudanças nas características dos receptores produzidas pela administração crônica de IMAOs se correlacionam melhor com a atividade antidepressiva do que o aumento na atividade do neurônio secundária ao aumento na concentração de neurotransmissores, e pode explicar a demora para início da ação terapêutica.6
Mais recentemente foram desenvolvidos IMAOs seletivos da MAO-A e da MAO-B, além de compostos reversíveis, que contornam o problema das crises hipertensivas (tabela 1). A moclobemida é um antidepressivo inibidor seletivo da MAO-A e reversível, que desamina 5-HT e NA, ao passo que inibidores seletivos da MAO-B, como a selegilina, não possuem ação antidepressiva significativa.6
Farmacocinética
Os IMAOs são bem absorvidos pelo trato gastrintestinal, sofrem biotransformação hepática rápida por oxidação e possivelmente têm metabólitos ativos. O início de ação se dá entre 7 a 10 dias com doses apropriadas em alguns pacientes, mas pode levar de 4 a 8 semanas para atingir o efeito terapêutico pleno. O pico de concentração plasmática é de 3 a 5 horas para isocarboxazida, 2 a 4 para fenelzina e 1 a 3,5 para tranilcipromina. Em média são necessários 10 dias para que a atividade da MAO se recupere, já que em 5 a 10 dias os IMAOs irreversíveis inibem as MAOs A e B de forma permanente. Elas voltam a ser produzidas em uma a duas semanas, mas nesta fase o paciente continua vulnerável ao desencadeamento de crises hipertensivas pelo aumento da concentração de aminas provenientes da dieta ou de medicamentos aminérgicos. A eficácia da fenelzina se correlaciona com a inibição de 80 % da MAO plaquetária, ao passo que o melhor preditor de resposta terapêutica da tranilcipromina parece ser a área sobre a curva cinética.6 A eliminação é renal, inclusive dos metabólitos.
A moclobemida inibe apenas a MAO A, por tempo menos prolongado (aproximadamente 24 horas apenas) e de forma reversível. Conseqüentemente, não é necessário aguardar duas semanas até que a MAO volte a ser produzida e outros antidepressivos possam ser prescritos.5,6
Efeitos colaterais
Os efeitos colaterais descritos a seguir foram selecionados de acordo com a relevância clínica.6
Necessidade de atenção médica
Freqüentes: hipotensão ortostática grave (vertigens e tonturas, especialmente ao levantar; podem ocorrer quedas); dividir ou reduzir as doses quando necessário.
Menos freqüentes: diarréia, edema nos pés e tornozelos (pode ceder espontaneamente em semanas); caso persista, monitorar eletrólitos para verificar a existência da síndrome de secreção inadequada do hormônio antidiurético; estimulação simpática (taquicardia e palpitação), menos freqüentemente nervosismo e excitação.
Raros: hepatite, leucopenia, síndrome de Parkinson, síndrome serotonérgica na combinação com medicamentos serotonérgicos (amitriptilina, clomipramina, doxepina, imipramina; fluoxetina, sertralina, paroxetina ou trazodona). A síndrome pode se manifestar por confusão mental, hipomania, inquietação, mioclonias, hiperreflexia, arrepios, calafrios, tremores, diarréia, incoordenação e febre. A melhora é rápida com a retirada das substâncias.
Necessidade de atenção médica se persistirem
Menos freqüentes: efeito anticolinérgico, síndrome da secreção inadequada do hormônio antidiurético (levando à diminuição na produção de urina); visão turva; estimulação do SNC (mioclonias durante o sono, inquietação ou agitação, dificuldades no sono) mais freqüente com tranilcipromina; disfunção sexual (anorgasmia em homens e mulheres, alterações ejaculatórias, raramente impotência masculina); sonolência (mais freqüente com fenelzina e isocarboxazida); cefaléia leve sem aumento da pressão arterial; aumento de apetite e peso relacionado à fissura por carboidratos; aumento da sudorese; hipotensão ortostática; vertigens, tontura, cansaço ou fraqueza leve; abalos musculares ou tremores.
Raros: anorexia; calafrios; constipação; boca seca.
Interações medicamentosas
Na tabela 2 estão descritas as principais interações entre IMAOs e outros medicamentos.7 Pelo fato de os IMAOs inibirem a MAO de forma permanente, é necessário adotar dieta pobre em tiramina, aminoácido precursor de catecolaminas, de modo a evitar uma crise hipertensiva potencialmente fatal.
Sintomas da crise hipertensiva
Cefaléia intensa, palpitações, dor torácica intensa, dilatação das pupilas, taquicardia ou bradicardia, aumento da fotossensibilidade, pode haver aumento da sudorese, febre ou sensação de frio, pele viscosa, náusea ou vômitos, rigidez de nuca. Existem relatos de hemorragia intracraniana (algumas vezes fatal) em conseqüência das crises hipertensivas. Palpitação ou cefaléia freqüente constituem sintomas prodrômicos da reação hipertensiva.
No anexo 1 anexo 1 encontra-se a lista de alimentos e medicamentos proibidos utilizada no Grupo de Doenças Afetivas do Instituto de Psiquiatria do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (Gruda-IPQ HCFMUSP).8
Antidepressivos Tricíclicos (ADTs)
Mecanismo de ação
O mecanismo de ação comum aos antidepressivos tricíclicos em nível pré-sináptico é o bloqueio de recaptura de monoaminas, principalmente norepinefrina (NE) e serotonina (5-HT), em menor proporção dopamina (DA). Aminas terciárias inibem preferencialmente a recaptura de 5-HT e secundárias a de NE (tabela 3). Atualmente se considera não haver diferenças significativas quanto à seletividade do bloqueio de recaptura pré-sináptico.3 A atividade pós-sináptica varia de acordo com o sistema neurotransmissor envolvido e geralmente é responsável pelos efeitos colaterais. Os ADTs bloqueiam receptores muscarínicos (colinérgicos), histaminérgicos de tipo 1, a2 e b-adrenérgicos, serotonérgicos diversos e mais raramente dopaminérgicos (tabela 3). Essas ações não se correlacionam necessariamente com efeito antidepressivo, mas com efeitos colaterais (tabela 4). O bloqueio do receptor 5-HT1 contribuiria para o efeito terapêutico.2
Contudo, esta ação aguda dos antidepressivos no sistema de transmissão monoaminérgica, por si só, não explicava a demora para o início da ação antidepressiva, observável clinicamente após duas semanas de uso. Estudos recentes das vias receptoras pós-sinápticas e de mensageiros secundários, assim como da expressão genética, podem desempenhar papel importante na elucidação das mudanças que ocorrem a longo prazo no funcionamento cerebral resultante da utilização crônica de antidepressivos.2
Embora o mecanismo de ação exato não tenha sido totalmente elucidado, sabe-se que os ADTs promovem agudamente aumento na eficiência da transmissão monoaminérgica (e possivelmente GABAérgica), envolvendo os sistemas noradrenérgico e serotoninérgico através do aumento na concentração sináptica de norepinefrina e serotonina por bloqueio de recaptura.3 Cronicamente os ADTs dessensibilizam receptores b1 adrenérgicos, serotonérgicos 5-HT2 e provavelmente 5-HT1A no sistema nervoso central. Sistemas mensageiros secundários estão envolvidos nessas mudanças.3 AMP cíclico, cálcio, diacilglicerol e fosfolípides estimulam a fosforilação de quinases proteicas, possivelmente envolvidas na síntese de catecolaminas. Podem aumentar a ligação de proteína G a receptores subseqüentemente dessensibilizados, exercendo ação reguladora no receptor. Os hormônios (como estradiol e progesterona) são substâncias também implicadas na alteração da sensibilidade ou no número de receptores pelos ADTs, interferindo na capacidade de ligação da imipramina ao hipotálamo.3
A ação antienurética do hidrocloridro de imipramina não está estabelecida. Acredita-se que esteja associada com o efeito anticolinérgico da imipramina. O efeito antiobsessivo da clomipramina talvez se correlacione com a inibição da recaptura de serotonina e conseqüente subsensiblização compensatória dos subtipos de receptores serotonérgicos. No transtorno do pânico, os estudos sugerem prejuízo no funcionamento do sistema nervoso autônomo, que causa liberação excessiva de norepinefrina do locus ceruleus. Pensa-se que os ADTs diminuam a taxa de disparo do locus ceruleus por regulação na função de receptores a2 e b-adrenérgicos e no turnover de noradrenalina. A ação antinevrálgica dos ADTs não está necessariamente relacionada à melhora da depressão. A analgesia pode ser mediada por mudanças na concentração central de monoaminas, particularmente serotonina, além do efeito direto ou indireto dos ADTs nos sistemas opióides endógenos. Na úlcera péptica, os ADTs são eficazes na melhora da dor e ajudam na cicatrização completa pela sua capacidade de bloquear receptores H2 nas células parietais e pelo efeito sedativo e anticolinérgico. Na bulimia nervosa parece haver efeito independente da melhora da depressão. O mecanismo de ação envolvido na incontinência urinária pode incluir atividade anticolinérgica, resultando no aumento da capacidade vesical, estimulação direta beta-adrenérgica e atividade agonista alfa- adrenérgica, resultando em aumento do tônus esfincteriano e também por bloqueio central de recaptação.9
Outras ações dos ADTs incluem efeito anticolinérgico periférico e central devido à potente e elevada afinidade de ligação por receptores muscarínicos; efeito sedativo pela forte afinidade de ligação por receptores histamina H1 e hipotensão ortostática devida a bloqueio alfa-adrenérgico. Além disto, os ADTs são agentes antiarrítmicos da classe 1A que, como a quinidina, em doses terapêuticas diminuem moderadamente a condução intraventricular e em doses elevadas podem causar bloqueio grave de condução e arritmias ventriculares.9
Farmacocinética
Os ADTs são bem absorvidos completamente pelo trato gastrintestinal, metabolizados em grande parte (55% a 80%) pelo efeito de primeira passagem, o pico plasmático é atingido mais rapidamente (1 a 3 horas) por aminas terciárias (como a amitriptilina) do que com aminas secundárias (desipramina e nortriptilina) que levam 4 a 8 horas para atingi-lo. São altamente lipofílicos, concentrando-se principalmente no miocárdio e em tecidos cerebrais, se ligam a proteínas plasmáticas e sofrem metabolismo primariamente hepático. Muitos ADTs apresentam farmacocinética linear, isto é, mudanças na dose levam a alteração proporcional no nível plasmático. A vida média de eliminação varia ( por exemplo, imipramina de 4 a 34 horas, amitriptilina de 10 a 46, clomipramina de 17 a 37 e nortriptilina de 13 a 88) e o estado de equilíbrio é atingido em cerca de 5 dias. A farmacocinética pode variar entre os sexos e a concentração pode diminuir antes da menstruação.5
Durante a gestação é possível utilizar ADTs, evitando-se preferencialmente no primeiro trimestre. Amitriptilina, clomipramina, desipramina e nortriptilina são antidepressivos que em estudos de reprodução em animais mostraram algum efeito adverso no feto e com os quais não há estudos adequados e bem controlados em humanos. Contudo, não há relatos de associação significativa entre ADTs e malformações congênitas descritos até o momento, mesmo no primeiro trimestre.5,9 Os ADTs devem ser suspensos duas semanas antes do parto, a fim de evitar problemas cardíacos, irritabilidade, desconforto respiratório, espasmos musculares, convulsões ou retenção urinária em neonatos.9
Mulheres lactantes podem tomar ADTs, preferencialmente imipramina e amitriptilina, mas também nortriptilina e clomipramina. A maprotilina deve ser evitada pela sua meia-vida longa.9
Efeitos colaterais
Anticolinérgicos: associados ao bloqueio muscarínico, são os mais freqüentes e sua intensidade declina com o passar do tempo ou redução do antidepressivo. São eles: boca seca (recomenda-se estimular higiene bucal freqüente), visão turva (por dificuldade de acomodação visual), obstipação (em idosos há risco de íleo paralítico) e retenção urinária.
Cardiovasculares: aumento da freqüência cardíaca, achatamento da onda T, raramente prolongamento do intervalo PR e aumento do complexo QRS, dose-dependentes e observados em concentrações plasmáticas acima dos níveis terapêuticos; hipotensão postural (idosos devem ser orientados e monitorados pelos riscos de quedas e nestes casos a nortriptilina estaria mais indicada);10 as propriedades antiarrítmicas quinidina-símile dos ADTs favorecem seu uso em pacientes com extrassístoles ventriculares.
Neurológicos: tremores de mãos, sedação (principalmente amitriptilina e maprotilina), latência para lembrar, mioclonias, parestesias, dificuldade para encontrar palavras e gagueira, agitação e hiperestimulação paradoxal, estados confusionais podem ocorrer em idosos, raramente convulsões (doses elevadas, aumento rápido, principalmente com maprotilina e clomipramina), movimentos coreoatetóides e acatisia. Os pacientes devem ser orientados para não operar máquinas perigosas, dirigir veículos, caso sonolentos, e evitar consumo de álcool.
Metabólicos e endócrinos: aumento da secreção de prolactina, mas galactorréia e amenorréia secundária são raras. Outro efeito raro é a hiponatremia da síndrome de secreção inadequada do hormônio antidiurético, descrita com amitriptilina e clomipramina.
Reações cutâneas: exantemas, urticária, eritema multiforme, dermatite esfoliativa e fotossensibilidade; ocorrem em 2% a 4% dos pacientes nas duas primeiras semanas de tratamento.
Gastrintestinais: raramente ocorrem alterações de função hepática.
Outros efeitos colaterais não menos importantes se referem àqueles que podem ser confundidos com a própria sintomatologia depressiva. Estão incluídos neste item: ganho de peso, associado ou não à preferência por carboidratos, principalmente com amitriptilina e imipramina; disfunções sexuais (redução da libido, retardo ou inibição ejaculatória e inibição do orgasmo); alterações do sono (pesadelos, alucinações hipnagógicas e hipnopômpicas). Aumento de ansiedade e "síndrome tricíclica precoce" podem ocorrer nos primeiros dias de tratamento, principalmente em pacientes com ataques de pânico, e melhoram com associação de benzodiazepínicos. Dificuldades de memória são mais comuns em idosos e no curso do tratamento profilático. Os efeitos colaterais dos ADTs e sua relação com o bloqueio de receptores estão resumidos na tabela 5.
Contra-indicações: Os ADTs estão contra-indicados no glaucoma de ângulo fechado. Efeitos na condução cardíaca normalmente não apresentam significado clínico, mas os ADTs são contra-indicados em bloqueios de ramo esquerdo, bloqueio AV total, alterações na condução intracardíaca e infarto agudo do miocárdio. O eletrocardiograma constitui um método sensível e deve ser solicitado quando se suspeita de alterações cardíacas e em pacientes acima de 50 anos.
Síndrome de abstinência ou de descontinuação: Em um pequeno grupo de pacientes a interrupção abrupta de ADTs, principalmente após tratamento prolongado, é acompanhada de uma síndrome de abstinência que ocorre nas primeiras 48 horas após a suspensão do antidepressivo. Os sintomas podem estar relacionados a um efeito rebote de hiperatividade colinérgica. Clinicamente a síndrome se caracteriza por sintomas de mal-estar geral, alterações gastrintestinais (náuseas, vômitos, diarréia), ansiedade, irritabilidade, insônia, sonhos vívidos, movimentos parkinsonianos ou acatisia. Podem ocorrer ataques de pânico, arritmias cardíacas, delirium e menos freqüentemente agitação. Recomenda-se a diminuição gradativa da medicação ao longo de algumas semanas. O esquema seguido no Gruda-IPq HCFMUSP consiste na retirada imediata de 50 % da dose e de 25 % a cada dois dias do restante.11
Intoxicação (superdosagem): Caracterizada por confusão, convulsões, alterações de concentração, sonolência grave, alargamento de pupilas, alteração da freqüência cardíaca, febre, alucinações, inquietação ou agitação, respiração curta ou difícil, cansaço e fraqueza intensa e vômitos. O tratamento da intoxicação consiste em diminuição da absorção (esvaziamento gástrico com lavagem), aumento da eliminação (administração de pasta de carvão ativado seguida de estimulação catártica), e tratamento específico das intercorrências cardiopulmonares.9
Interações medicamentosas
Interações medicamentosas de significativa importância entre antidepressivos tricíclicos e outros medicamentos comumente utilizados em idosos:7
Analgésicos: os ADTs possuem efeito antiálgico, permitindo que doses menores de analgésicos sejam empregadas.
Anestésicos: a administração de halotano e pancurônio requer cautela pelo efeito anticolinérgico dos ADTs; recomenda-se o uso de relaxantes musculares sem efeitos vagolíticos e simpatomiméticos.
Agentes anticolinérgicos: a administração conjunta de ADTs e antiparkinsonianos pode levar à potencialização de efeitos atropínicos. Sintomas de síndrome anticolinérgica podem ocorrer, tais como: ansiedade, agitação, desorientação, disartria, comprometimento de memória, alucinações, mioclonias, convulsões, taquicardia, arritmias, midríase, elevação da temperatura corporal, obstipação intestinal e retenção urinária.
Anticoagulantes: relatos de casos isolados sugerem cuidados na interação entre anticoagulantes e antidepressivos tricíclicos, especialmente no que se refere à análise do tempo de protrombina em pacientes que recebem tratamentos combinados.
Anticonvulsivantes: a carbamazepina pode aumentar o metabolismo de imipramina, doxepina e amitriptilina, reduzindo em 42% a 50 % os níveis plasmáticos; ADTs reduzem o limiar convulsígeno e podem comprometer o efeito de barbitúricos; os níveis plasmáticos da fenitoína podem ser elevados pela imipramina, mas não por nortriptilina ou amitriptilina.
Anti-hipertensivos: a guanetidina não deve ser utilizada em pacientes que fizerem uso de antidepressivos bloqueadores de recaptação de norepinefrina; a clonidina também deve ser evitada; metildopa e diuréticos tiazídicos podem ser empregados, evitando hipotensão e hipocalemia; verapamil e diltiazem podem inibir a metabolização de imipramina por interação no sistema citocromo P450, podendo ser necessário reduzir a dose do antidepressivo.
Bloqueadores histamínicos H2: a cimetidina pode inibir a metabolização hepática de ADTs, elevando níveis séricos e risco de toxicidade; ela pode aumentar a biodisponibilidade de imipramina, porém não de nortriptilina; suspender a cimetidina do paciente em uso crônico de ADTs pode reduzir os níveis séricos terapêuticos; sugere-se monitoração plasmática ao introduzir e retirar cimetidina.
Levodopa: a associação pode ter efeito sinérgico nos sistemas colinérgicos e catecolaminérgicos, aumentando efeitos colaterais.
Quinidina: associação com desipramina pode aumentar os níveis séricos e o risco de toxicidade.
Reserpina: a reserpina depleta agudamente monoaminas intraneuronais; a associação pode levar a efeitos colaterais como diarréia, vasodilatação cutânea ou mesmo sintomas maniformes; recomenda-se cuidado nesta combinação.
Aminas simpatomiméticas: a administração de noradrenalina ou outras aminas simpatomiméticas em pacientes recebendo ADTs pode levar a efeito sinérgico, aumentando o tônus simpático.
Antidepressivos inibidores seletivos da recaptação de serotonina (ISRSs)
Os ISRSs, citalopram, fluoxetina, fluvoxamina, paroxetina e sertralina são o resultado de pesquisa racional para encontrar medicamentos tão eficazes quanto os ADTs, mas com poucos problemas de tolerabilidade e segurança. Os ISRSs inibem de forma potente e seletiva a recaptação de serotonina, resultando em potencialização da neurotransmissão serotonérgica. Embora compartilhem o principal mecanismo de ação, os ISRS são estruturalmente distintos com marcadas diferenças no perfil farmacodinâmico e farmacocinético. A potência da inibição de recaptação da serotonina é variada, assim como a seletividade por noradrenalina e dopamina. Sertralina e paroxetina são os mais potentes inibidores de recaptação.12 A potência relativa da sertralina em inibir a recaptação de dopamina a diferencia farmacologicamente dos outros ISRSs. A afinidade por neuroreceptores, tais como sigmal, muscarínicos e 5-HT2c, também difere muito. Mais ainda, a inibição da sintetase óxido-nítrica pela paroxetina e possivelmente por outros ISRSs, pode ter efeitos farmacodinâmicos significativos. Citalopram e fluoxetina são misturas racêmicas de diferentes formas quiral que possuem perfis farmacodinâmico e farmacocinético variados. Fluoxetina possui metabólito de ação prolongada e farmacologicamente ativo. Os ISRSs também possuem perfis farmacocinéticos variados, que incluem meia vida, farmacocinética linear versus não linear, efeito da idade na sua depuração e no seu potencial de inibir isoenzimas metabolizadoras de medicamentos do citocromo P450 (CYP). Estas diferenças farmacológicas e farmacocinéticas sustentam as diferenças clínicas cada vez mais importantes dos ISRSs.12
Farmacocinética
Embora todos os ISRSs apresentem o mesmo mecanismo de ação, as diferenças entre as estruturas moleculares fazem com que os diferentes compostos apresentem perfis farmacocinéticos diversos (tabela 5). Todos os ISRSs apresentam alta ligação proteica (fluvoxamina e citalopram em menor grau). A fluoxetina é a única que apresenta metabólito com atividade clínica significativa (inibição da recaptação de serotonina e inibição de isoenzimas do citocromo P 450) , a norfluoxetina. A meia-vida prolongada da fluoxetina e da norfluoxetina e o tempo necessário para se atingir o estado de equilíbrio apresentam significado clínico, como a maior latência para o início da ação antidepressiva.12 As concentrações plamáticas de sertralina e citalopram são proporcionais às doses administradas (farmacocinética linear), o que não ocorre com fluoxetina, paroxetina e fluvoxamina, cuja farmacocinética não é linear. Estes ISRSs diminuem seu metabolismo por ação inibitória dose-dependente das isoenzimas do citocromo P450 (CYP), o que significa que aumentos na dose administrada de fluoxetina, paroxetina e fluvoxamina levam a aumentos desproporcionais nos níveis plasmáticos, meias-vidas e possivelmente efeitos colaterais.12 Os ISRSs são rapidamente absorvidos, sofrem menos efeito do metabolismo de primeira passagem, se ligam fortemente a proteínas plasmáticas, todos (em menor proporção fluvoxamina) deslocam outras drogas da ligação proteica, aumentando seu nível plasmático. Metabolizados primariamente pelo fígado, todos os ISRSs afetam as enzimas metabolizadoras do citocromo P-450 (em menor proporção sertralina) e podem comprometer o metabolismo de outras drogas metabolizadas por este sistema. Tem-se demonstrado que fluoxetina e paroxetina diminuem seu metabolismo com o tempo. O pico plasmático da sertralina aumenta 30% quando o medicamento é ingerido com alimentos, pela diminuição do metabolismo de primeira passagem.
Efeitos colaterais
Em função de sua ação seletiva, apresentam perfil mais tolerável de efeitos colaterais, existindo também diferenças entre os principais efeitos colaterais dos diferentes ISRSs. De forma geral, os efeitos colaterais mais freqüentemente relatados são: gastrintestinais (náuseas, vômitos, dor abdominal, diarréia), psiquiátricos (agitação, ansiedade, insônia, ciclagem para mania, nervosismo), alterações do sono, fadiga, efeitos neurológicos (tremores, efeitos extrapiramidais), perda ou ganho de peso, disfunções sexuais, reações dermatológicas.13
Gastrintestinais: os efeitos anticolinérgicos da paroxetina podem levar a maior incidência de obstipação intestinal em detrimento de diarréia; por outro lado, alguns estudos sugerem maior incidência de diarréia com a sertralina em relação à fluoxetina, e citalopram.13
Reações dermatológicas: mais freqüentes com a fluoxetina. Freqüentemente aparecem na forma de urticária, que pode estar acompanhada por febre, artralgia e eosinofilia.13
Efeitos psiquiátricos: a fluoxetina parece estar mais relacionada ao aparecimento de efeitos colaterais como agitação, insônia, ansiedade, ciclagem para a mania e nervosismo. Entretanto, os outros ISRSs podem apresentar os mesmos efeitos com o emprego de doses mais elevadas.
Alterações de peso: a sertralina está associada a uma discreta perda de peso no início do tratamento; a fluoxetina parece ser mais potente na inibição do apetite, com maior perda de peso no início do tratamento; a paroxetina, ao contrário, foi associada a ganho de peso, o que também foi relatado com o citalopram.
Disfunção sexual: o uso de ISRSs foi associado principalmente a retardo ejaculatório em homens e anorgasmia em mulheres; a paroxetina está associada a uma maior incidência desses efeitos colaterais, o que poderia ser explicado por sua potência na inibição da recaptação de serotonina e sua mínima atividade dopaminérgica.
Síndrome serotonérgica: A seletividade de ação dos ISRSs, que explica a redução no perfil de efeitos colaterais, pode, por outro lado, aumentar o risco de interação com outras substâncias que afetem a transmissão serotonérgica, levando ao aparecimento de sintomas que são freqüentemente descritos como síndrome serotonérgica. Os sintomas podem aparecer em casos de intoxicação por ISRSs ou mesmo com o emprego de doses terapêuticas de substâncias serotonérgicas associadas, como ADTs, IMAOs, ISRSs e lítio. Também podem ocorrer na substituição entre substâncias, quando não se observa período de wash-out adequado para a total eliminação da substância. Foram descritos: alterações cognitivas e comportamentais (confusão, hipomania, agitação), do sistema nervoso autônomo (diarréia, febre, diaforese, efeitos na pressão arterial, náuseas e vômitos) e neuro-musculares (mioclonias, hiperreflexia, incoordenação e tremores).
Sintomas de retirada (descontinuação): Os sintomas que aparecem na retirada dos ISRSs são clinicamente benignos, podem aparecer dentro de 1 a 10 dias após a retirada da medicação (embora no caso da fluoxetina possam aparecer várias semanas depois, em função de seu perfil farmacocinético.), e persistir por até 3 semanas. Os sintomas mais freqüentes são tonturas, vertigens, ataxia, sintomas gastrintestinais (náuseas e vômitos), sintomas gripais, distúrbios sensoriais (parestesias), alterações de sono (insônia, sonhos vívidos), e sintomas psíquicos (irritabilidade, agitação, ansiedade). Assim como acontece com outras substâncias psicoativas, estes sintomas podem ser o resultado de alterações adaptativas que mais freqüentemente envolvem o ajustamento de receptores para compensar a atividade farmacológica da droga (efeito rebote). O aparecimento dos sintomas correlaciona-se com a queda nos níveis plasmáticos dos ISRSs, o que explica sua maior incidência na retirada de paroxetina e fluvoxamina do que durante a retirada de fluoxetina. Entretanto, a maior ocorrência desses sintomas com a paroxetina pode ser explicada não apenas pelo perfil farmacocinético, mas também por seus efeitos anticolinérgicos.13
Cuidados a considerar em casos especiais
Uso na gestação e lactação: não há evidências de teratogenicidade em seres humanos. Foi relatada maior incidência de partos prematuros com o uso de ISRSs no terceiro trimestre de gestação. Sintomas de abstinência em neonatos, tais como tremores, irritabilidade, inquietação e nervosismo puderam ser observados (no caso da fluoxetina estão relacionados a níveis plasmáticos de fluoxetina e norfluoxetina). A fluoxetina e o citalopram são distribuídos para o leite materno em níveis terapêuticos e seu uso em lactantes não é recomendado. O lactente pode receber até 17% da dose materna de fluoxetina; com sertralina, paroxetina e fluvoxamina as concentrações encontradas são muito pequenas. Entretanto, deve-se pesar individualmente a relação risco/benefício de seu uso.5,9 Para os demais ISRSs não há estudos conclusivos em humanos.
Uso em hepatopatas: a redução na metabolização dos ISRSs em pacientes com comprometimento hepático pode implicar na necessidade de usar doses mais baixas. Pacientes com cirrose hepática alcoólica apresentam depuração reduzida de fluoxetina, sertralina e fluvoxamina.5
Interações medicamentosas
Assim como o perfil farmacocinético, o potencial para interações medicamentosas difere entre os vários ISRSs. O principal mecanismo das interações medicamentosas dos ISRSs envolve a inibição de diferentes isoenzimas do citocromo P450: CYP2D6, CYP3A3/4, CYP1A2, CYP2C9/10 e CYP2C195 (tabela 6).
Além dos ISRSs, outros compostos antidepressivos foram desenvolvidos e estudados a partir da década de 1980. Os antidepressivos de nova geração apresentam um perfil reduzido de efeitos colaterais e interações medicamentosas, constituindo importantes opções no tratamento de episódios depressivos. Entre os antidepressivos de nova geração, descritos a seguir, estão incluídos: venlafaxina, nefazodona, trazodona, reboxetina, bupropion e mirtazapina.
Inibidor seletivo de recaptura de 5-HT/NE (ISRSN)
Venlafaxina
Mecanismo de ação
A venlafaxina e seu metabólito ativo O-desmetilvenlafaxina (ODV) são inibidores seletivos da recaptação de serotonina e noradrenalina (ISRSNs), e apresentam fraca atividade como inibidores da recaptação de dopamina (clinicamente significativo apenas com doses elevadas). A potência da inibição de recaptura de serotonina é algo superior à de recaptura de noradrenalina, ocorrendo em doses inferiores. A venlafaxina e o ODV não apresentam afinidade por receptores adrenérgicos alfa-1, receptores muscarínicos ou histamínicos e também não inibem a monoamino-oxidase. Alteram a sensibilidade de receptores beta-adrenérgicos após dose única, diferente de outros antidepressivos que levam à dessensibilização desses receptores após doses repetidas.14, 15
Farmacocinética
A venlafaxina é rapidamente absorvida, sua biodisponibinidade é de 45%, e a ingestão com alimentos retarda, porém não compromete sua absorção. A liberação da venlafaxina da formulação de liberação prolongada (XR) é controlada pela membrana e independe do pH. Embora a absorção da formulação de liberação prolongada ocorra em ritmo mais lento e em concentrações plasmáticas inferiores, o total absorvido é o mesmo. A ligação proteica é moderada (cerca de 30% para a venlafaxina e 40% para o ODV). A venlafaxina sofre metabolização hepática com importante efeito de primeira passagem. O ODV é formado por 0-desmetilação pela isoenzima 2D6 do citocromo P450 (CYP2D6). Estudos in vitro evidenciam o envolvimento do CYP3A4 na metabolização da venlafaxina na N-desmetil-venlafaxina (menos ativo que o ODV).
Efeitos colaterais
Os efeitos colaterais mais freqüentemente relatados com o uso da venlafaxina são: náuseas, tonturas, sonolência; com doses acima de 225 mg/dia podem aparecer sintomas como hipertensão, sudorese abundante, tremores.14 A hipertensão aparece como resultado da inibição da recaptação de noradrenalina, desenvolvendo-se em cerca de 3% dos pacientes que fazem uso de 100 mg/dia; 5% dos pacientes em uso de doses entre 101 e 200 mg/dia; 7% dos pacientes em uso de doses entre 201 e 300 mg/dia; e 13% dos pacientes em uso de doses acima de 300 mg/dia. Porém, em menos de 1% dos pacientes o tratamento deve ser interrompido por este motivo.9 A magnitude do aumento nos níveis da pressão arterial é de 2 mm/Hg com doses de 225 mg/dia e de 7,5 mm/Hg com doses de 375 mg/dia. O tratamento da hipertensão, quando necessário, inclui o uso de drogas antidepressivas.14 Os efeitos colaterais na esfera sexual aparentam ser dose-dependentes e parece não haver desenvolvimento de tolerância. Podem ser relatados diminuição da libido, anorgasmia, retardo ejaculatório e impotência.9
Cuidados a considerar em casos especiais
Não há estudos controlados em gestantes. Estudos com ratos e coelhos empregando doses superiores às terapêuticas em até 12 vezes não evidenciaram efeitos teratogênicos. Não se sabe se a venlafaxina é excretada no leite materno, porém não foram relatados problemas a este respeito. O uso em pacientes com doenças cardiovasculaes e hipertensão deve ser bem avaliado, uma vez que a indução de elevação nos níveis da pressão arterial ou a hipotensão postural podem agravar condições pré-existentes. O metabolismo da venlafaxina está alterado em pacientes com comprometimento hepático e deve-se considerar redução nas doses em até 50% no caso de comprometimento hepático grave ou moderado. A excreção da venlafaxina pode ser alterada em pacientes com comprometimento renal. Pacientes com comprometimento leve ou moderado devem receber 25% a 50% da dose. Pacientes em hemodiálise devem receber 50% da dose, que deve ser administrada após a sessão de diálise.
Interações medicamentosas
As interações medicamentosas de significado clínico estão descritas na tabela 7.
Inibidores de recaptura de serotonina e antagonista alfa 2 (IRSAs)
Nefazodona
Mecanismo de ação
A nefazodona, uma fenilpiperazina, embora estrutural e quimicamente relacionada à trazodona, difere desta farmacologicamente. O mecanismo de ação da nefazodona se dá por meio da inibição da captação neuronal de serotonina e noradrenalina. É antagonista de receptores 5-HT2 e de receptores alfa-1 adrenérgicos.9 A administração crônica de nefazodona leva à dessensibilização de receptores 5-HT2a, porém não de receptores beta-adrenérgicos, sugerindo que não iniba a captação de noradrenalina in vivo.14 Estudos in vitro não evidenciaram afinidade significativa por receptores alfa-2 adrenérgicos, 5-HT1a, colinérgicos, dopaminérgicos, histamínicos, benzodiazepínicos, GABAérgicos.14, 9 Promove subsensibilização de receptores beta- adrenérgicos.5
Farmacocinética
A nefazodona é absorvida rapidamente; a ingestão com alimentos retarda a absorção e reduz a biodisponibilidade em cerca de 20%. Sua biodisponibilidade ab- soluta é baixa (20% da dose oral) e variável, em função do elevado metabolismo. A nefazodona apresenta elevada distribuição tissular (volume de distribuição entre 0,22 e 0,87 l/kg) e ligação proteica (99%). A biotransformação hepática fornece três metabólitos ativos identificados: a hidroxinefazodona (OH-NEF), que tem perfil farmacológico semelhante ao da nefazodona; a triazolodiona, com perfil similar, porém menor afinidade pelo receptor 5-HT2a; e a meta-clorofenilpiperazina (mCPP), semelhante à nefazodona e também agonista de receptores serotonérgicos 5-HT2c. A meia-vida da nefazodona é dose-dependente e varia de duas a quatro horas; os picos plasmáticos são atingidos em cerca de uma hora e o estado de equilíbrio em cinco dias (55% dos pacientes sofrem excreção renal e 20% a 30%, excreção fecal).5,9
Efeitos colaterais
Os efeitos colaterais de relevância clínica mais freqüentemente relatados são: cefaléia, boca seca, sonolência, náuseas, obstipação intestinal e ataxia; também foram relatados turvação de visão, dispepsia, fraqueza e "rash" cutâneo 9,16,17. Os efeitos cardiovasculares da nefazodona descritos em estudos realizados na fase anterior à comercialização incluem a queda nos níveis de pressão arterial (5,1% dos pacientes), hipotensão postural (2,8% dos pacientes) e bradicardia (1,5% dos pacientes).17
Intoxicação: Os sintomas agudos da intoxicação pela nefazodona incluem hipotensão, náuseas, vômitos e sonolência excessiva. O tratamento consiste em medidas de suporte e tratamento sintomático. A lavagem gástrica pode ser útil para reduzir a absorção da nefazodona.
Cuidados a considerar em casos especiais
Gestação e lactação: não há estudos controlados sobre o uso da nefazodona em mulheres gestantes. Em estudos com animais, não se observou o desenvolvimento de malformações atribuíveis à nefazodona em coelhos e ratos expostos a doses cinco a seis vezes superiores às doses recomendadas.9 Orienta-se evitar seu uso no primeiro trimestre de gravidez. Também não é conhecida a distribuição da nefazodona e seus metabólitos no leite materno. A Academia Americana de Pediatria classifica os IRSAs como medicamentos em que seus efeitos na amamentação são desconhecidos, mas podem ser relevantes.5
Uso em pacientes com doenças cardiovasculares, cerebrovasculares e em uso de drogas anti-hipertensivas: em função dos efeitos cardiovasculares da nefazodona, recomenda-se cautela nestes pacientes.9
Uso em idosos: iniciar com doses menores; idosos podem levar mais tempo para responder (até 12 semanas). Deve-se monitorar a ação no SNC e anticolinérgica, tendo cuidado com a interação de outros medicamentos com ação semelhante devido ao risco de efeito somatório (confusão, desorientação, delirium). Idosos são mais sensíveis a efeitos anticolinérgicos, ao risco de quedas por hipotensão e a prejuízo cognitivo.5
Interações medicamentosas
A nefazodona é um inibidor in vitro do citocromo P450 3A4 e a administração de drogas metabolizadas por essa isoenzima requer cautela. É fraco inibidor in vitro da isoenzima 2D6 e não altera in vitro a isoenzima 1A2. As principais interações medicamentosas da nefazodona estão descritas na tabela 8.5
Trazodona
Mecanismo de ação
O mecanismo de ação postulado para a trazodona envolve a inibição da recaptação de serotonina e noradrenalina.15 A longo prazo ocorre a dessensibilização e diminuição no número de receptores beta-adrenérgicos e 5-HT2A. Apresenta atividade antagonista de receptores alfa-1-adrenérgicos e anti-histamínicos, mais relacionadas aos seus efeitos colaterais. O metabólito ativo mCPP também apresenta algum grau de atividade serotonérgica pós-sináptica.9,17
Farmacocinética
A trazodona é bem absorvida pelo trato gastrintestinal e se ingerida às refeições, ou imediatamente após , pode haver aumento na quantidade absorvida, redução na concentração máxima e aumento no tempo necessário para atingir pico plasmático. Em geral os picos plasmáticos são atingidos em duas horas. Apresenta alta ligação proteica (cerca de 90%) e sofre hidroxilação hepática. Sua meia-vida é de 6 a 11 horas. A eliminação é renal (75%, predominantemente como metabólitos inativos) e biliar (20%). Seu metabólito ativo é o mCPP.9
Efeitos colaterais
Os efeitos colaterais mais freqüentes da trazodona são: sedação, hipotensão ortostática, tonturas, cefaléia, náuseas, boca seca. Reações alérgicas e irritação gástrica podem aparecer. Alguns relatos de casos sugerem associação entre a trazodona e o aparecimento de arritmias em pacientes que já apresentavam contrações ventriculares prematuras ou prolapso de válvula mitral. A trazodona está associada à ocorrência de priapismo (ereção peniana prolongada na ausência de estímulo). Neste caso, deve-se suspendê-la. Sugere-se avaliar com o paciente a troca do antidepressivo caso perceba que a freqüência e a duração das ereções está aumentando. O tratamento do priapismo consiste na injeção intra-cavernosa de solução de epinefrina (1 mcg/ml). Outras disfunções sexuais também podem aparecer.9,17
Intoxicação: Casos de intoxicação por trazodona freqüentemente se caracterizam por apresentar sedação, hipotensão, perda de coordenação muscular, náuseas e vômitos. O tratamento consiste na redução da absorção com lavagem gástrica e administração de carvão ativado, na tentativa de aumento da eliminação com diurese forçada e na adoção de medidas de monitorização cardíaca e de suporte.17
Cuidados a considerar em casos especiais
O uso da trazodona na gestação está contra-indicado; estudos animais associam o uso de trazodona a malformações fetais.9 A trazodona é excretada no leite materno e o aleitamento é contra-indicado. Em pacientes com comprometimento hepático e/ou renal seu uso deve ser feito com cautela, em função de alterações no metabolismo e excreção da droga.
Interações medicamentosas
As interações medicamentosas clinicamente significativas envolvem substâncias depressoras do SNC e IMAOs, e estão descritas na tabela 8.
Inibidor seletivo de recaptação de norepinefrina (ISRN)
Reboxetina
A reboxetina é o primeiro composto comercializado de uma nova classe de antidepressivos inibidores da recaptação de noradrenalina (IRNAs) , estruturalmente semelhantes à viloxazina.18 Apresenta atividade seletiva sobre a recaptação de noradrenalina, com atividade antagonista alfa-2. Não possui efeitos significativos sobre receptores colinérgicos, histamínicos, alfa-1-adrenérgicos, ou na inibição da monoaminoxidase e seu efeito antidepressivo foi descrito inicialmente na década de 1980.18,19
Farmacocinética
A reboxetina é absorvida pelo trato gastrintestinal, apresenta alta ligação à glicoproteína alfa-1 plasmática. Atinge pico plasmático em 1,5 a 2,5 horas. Sofre metabolização hepática por hidroxilação e oxidação principalmente. Não interage com isoenzimas do sistema citocromo P450. Sua meia-vida é de cerca de 12 a 13 horas e a eliminação se dá pela urina (76% na forma inalterada e metabólitos) e fezes (7% a 16%).18,20
Efeitos colaterais
Os efeitos colaterais mais significativos da reboxetina são: taquicardia, impotência, hesitação ou retenção urinária, insônia, sudorese excessiva, obstipação intestinal, boca seca. Em geral estes efeitos têm intensidade moderada, mesmo em doses acima de 8 mg/dia.21
Cuidados a considerar em casos especiais
Quanto ao uso na gestação e lactação ainda não há dados. Em pacientes cardiovasculares a reboxetina pode levar a um aumento de freqüência cardíaca e a um leve decréscimo na pressão arterial. Deve ser usada com precaução. Em pacientes idosos não há estudos. Entretanto, pacientes com hipertrofia prostática podem sentir-se especialmente incomodados pela retenção urinária. A meia-vida prolongada requer ajuste de doses, ainda sem diretrizes definidas. A extensa metabolização hepática sofrida pela reboxetina sugere a necessidade de ajustes de doses em pacientes hepatopatas, porém neste caso também não há condutas definidas.21
Interações medicamentosas
A ausência de interação com as enzimas do citocromo P450 e a seletividade da ação conferem à reboxetina baixo potencial de interações medicamentosas.18 Embora ainda não existam estudos definitivos sobre o assunto, os já realizados demonstraram que a reboxetina não apresenta interação farmacocinética com o lorazepam em indivíduos saudáveis e também não se observou interação farmacodinâmica (desempenho psicomotor ou cognitivo) com o álcool em estudo duplo-cego que incluiu número reduzido de pacientes.20
Inibidor seletivo de recaptura de dopamina (ISRD)
Bupropion
Mecanismo de ação
Embora não completamente conhecido, o mecanismo de ação do bupropion se dá através de sua atividade noradrenérgica e dopaminérgica. O bupropion aumenta a liberação de noradrenalina corpórea15 e é um fraco inibidor in vitro da captação neuronal de noradrenalina e de dopamina,9 porém de relevância farmacológica.14 O hidroxibupropion é seu metabólito ativo.9 O bupropion não inibe a monoaminoxidase e tem pouca afinidade pelo sistema serotonérgico.9,14,15 Também não interage com receptores histamínicos e colinérgicos, levando a uma maior tolerabilidade.14,15
Farmacocinética
O bupropion é rapidamente absorvido pelo trato intestinal, porém o metabolismo pré-sistêmico elevado diminui sua biodisponibilidade. O bupropion e o hidroxibupropion apresentam alta ligação proteica (84% e 77%, respectivamente). O bupropion cruza rapidamente a barreira hematoencefálica e a placenta, sendo distribuído no leite materno. É extensivamente metabolizado (inclusive metabolização pré-sistêmica) e três de seus metabólitos apresentam alguma atividade, segundo estudos em animais: o hidroxibupropion (formado principalmente pelo citocromo P450 2B6), com potência equivalente ao bupropion, o treoidrobupropion e o eritroidrobupropion, formados por hidroxilação e/ou redução e que apresentam de 1/10 a 1/2 da potência do bupropion. Sua meia-vida de distribuição é de cerca de 3 a 4 horas, a meia-vida de eliminação após dose única é de 14 horas e, no estado de equilíbrio, de cerca de 21 horas (podendo variar entre 12 e 30 horas). A meia-vida de eliminação do hidroxibupropion é de cerca de 20 horas. Os picos plasmáticos do bupropion e do hidroxibupropion são de 1,5 e 3 horas, respectivamente, pasando para 3 e 6 horas usando a formulação de liberação prolongada. A eliminação renal é de 1% na forma inalterada, acima de 60% como metabólitos em 24 horas e acima de 80% em 96 horas; a eliminação fecal é de 10%, principalmente na forma de metabólitos.9
Efeitos colaterais
O bupropion apresenta boa tolerabilidade. Entre os antidepressivos de nova geração, apresenta o menor potencial de indução de efeitos colaterais e a menor incidência de descontinuação do tratamento por intolerância.16 Os efeitos colaterais mais freqüentemente observados são agitação, ansiedade, rash cutâneo, diminuição do apetite, boca seca e obstipação intestinal. Entretanto, o aumento do risco de indução de convulsões é maior que o de outros antidepressivos, e mais freqüente com doses elevadas. A incidência de convulsões com a forma de liberação prolongada é de 0,1% em doses até 300 mg/ dia e de 0,4% em doses acima de 400 mg/ dia. Com o uso da forma de liberação imediata o risco passa para 0,4% com doses entre 300 e 450 mg/dia, podendo aumentar até dez vezes em doses entre 450 e 600 mg/dia.9 Para minimizar o risco de convulsões recomenda-se que cada dose do composto de liberação imediata não exceda 150 mg e do composto de liberação prolongada não exceda 200 mg. Deve-se observar intervalo de 4 horas entre as doses do composto de liberação imediata e de 8 horas entre as tomadas do composto de liberação prolongada.
Intoxicação: Os efeitos clínicos da ingestão de doses elevadas de bupropion são: alucinações, diminuição do nível de consciência, náuseas, vômitos, convulsões (em 1/3 dos casos) e taquicardia, que pode evoluir para bradicardia e assistolia.9 O tratamento da intoxicação inclui medidas para diminuição da absorção. Pacientes estuporosos ou comatosos devem ser entubados; em seguida realizar lavagem gástrica e administração de carvão ativado a cada 6 horas se a ingestão ocorreu nas últimas 12 horas. Não se recomenda o xarope de ipeca para induzir vômitos pelo risco de convulsões. No caso de convulsões, administrar benzodiazepínicos por via endovenosa. É fundamental monitorizar ECG e EEG por pelo menos 48 horas e equilíbrio eletrolítico e ácido-básico em pacientes com estado de mal epilético. Medidas gerais de suporte, como diurese forçada, diálise ou hemoperfusão não são indicados, pois o bupropion e seus metabólitos apresentam lenta difusão dos tecidos para o plasma.9
Cuidados a considerar em casos especiais
Estudos adequados e bem controlados em humanos ainda não foram realizados, porém sabe-se que o bupropion passa rapidamente à placenta. Seu uso na gestação não é recomendado.17 O bupropion é distribuído para o leite materno, oferecendo risco potencial (como convulsões, por exemplo) para o lactente. Em pacientes geriátricos acima de 60 anos não há limitações ao uso de bupropion, mas alterações metabólicas relacionadas à idade podem causar intolerância aos efeitos colaterais, e alterações renais e hepáticas podem exigir redução nas doses prescritas. Em pacientes com história de traumatismo craniano, tumores cerebrais, quadros cerebrais orgânicos ou alterações eletroencefalográficas, o uso do bupropion não é recomendado em função do risco de convulsões.17 Em doentes renais ou hepatopatas a metabolização e a excreção do bupropion podem estar alteradas. Sugere-se iniciar o tratamento com baixas doses e monitorar intensamente o paciente. Na anorexia e bulimia não é recomendado pelo maior risco de convulsões.9
Interações medicamentosas
As interações medicamentosas clinicamente significativas estão descritas na tabela 9.
Antidepressivo noradrenérgico e específico serotoninérgico (ANES)
Mirtazapina
Mecanismo de ação
A ação da mirtazapina se dá através do aumento da atividade noradrenérgica e serotonérgica central. A mirtazapina é um antagonista de auto e hetero-receptores alfa-2 adrenérgicos pré-sinápticos e antagonista 5-HT2 e 5-HT3 pós-sináptico. Apresenta fraca afinidade pelos receptores 5-HT1a e 5-HT1b pós-sinápticos. Sua afinidade pelos receptores histamínicos H1 explica o efeito sedativo. Apresenta fraca atividade por receptores muscarínicos e dopaminérgicos.15,22
Farmacocinética
A mirtazapina é bem absorvida pelo trato gastrintestinal, porém devido ao metabolismo de primeira passagem sua biodisponibilidade é de 50%. Apresenta alta ligação a proteínas plasmáticas (85%). Os picos plasmáticos são atingidos em cerca de duas horas e o estado de equilíbrio em cinco dias, apresentando relação linear com a dose ingerida. A mirtazapina sofre metabolização hepática, principalmente desmetilação e hidroxilação seguida de conjugação ao ácido glucurônico. Seus metabólitos são ativos, encontrados em níveis baixos. A meia-vida de eliminação é de 20 a 40 horas (mais longa em mulheres de todas as idades). Os metabólitos são eliminados na urina (75%) e nas fezes (15%).9,22
Efeitos colaterais
A mirtazapina apresenta boa tolerabilidade. Os efeitos colaterais mais freqüentemente relatados são: sedação excessiva, ganho de peso (principalmente com o uso de doses baixas), boca seca, edema, obstipação intestinal, dispnéia. Em estudos clínicos realizados antes de seu lançamento observou-se a ocorrência de 2 casos (entre 2.796 pacientes) de agranulocitose reversível e de um caso de neutropenia grave também reversível.22 Sugere-se que a mirtazapina seja suspensa em pacientes que apresentarem febre ou outros sinais de infecção e tiverem baixa contagem de leucócitos.9
Intoxicação: A mirtazapina apresenta alguma segurança em casos de intoxicação (relato de ingestão de até 30 vezes a dose recomendada), sendo mais segura do que a imipramina.22 Os sinais e sintomas presentes em casos de intoxicação por mirtazapina incluem desorientação, tonturas, comprometimento de memória, taquicardia, sedação excessiva.9,22 O tratamento inclui medidas de suporte geral e monitorização das funções vitais. Pode-se empregar medidas para reduzir a absorção, como indução de emese e lavagem gástrica seguida da administração de carvão ativado.
Cuidados a considerar em casos especiais
Ainda não existem estudos controlados sobre o uso da mirtazapina na gestação em humanos. Sugere-se evitar o uso na gestação. Não se sabe se a mirtazapina é distribuída para o leite materno. A diminuição da eliminação da mirtazapina em idosos (40% entre homens e 10% entre mulheres) e a diminuição da função renal podem exigir ajustes de dose. Recomenda-se iniciar com 7,5mg ao dia e aumentar a dose para 15mg em uma a duas semanas dependendo da resposta e dos efeitos colaterais e monitorar sedação e efeito anticolinérgico.5 Foram observadas elevações nas transaminases hepáticas superiores a três vezes o valor normal em indivíduos sem comprometimento hepático sob uso de mirtazapina, sem o desenvolvimento de sinais e sintomas de alteração da função hepática, que retornaram aos parâmetros normais com a suspensão da droga. Pacientes com comprometimento da função hepática apresentam diminuição de 30% da depuração após ingestão de dose única de 15mg.9
Interações medicamentosas
As principais interações medicamentosas da mirtazapina estão sintetizadas na tabela 10.
Conclusão
O avanço da pesquisa em psicofarmacologia de antidepressivos vem oferecendo aos pacientes substâncias com perfis farmacocinéticos, de tolerância e de interações com outras drogas bastante diferentes entre si. Apesar disto, os mecanismos de ação propostos para cada um deles permanecem vinculados às teorias monoaminérgica, de aumento da oferta de neurotransmissores na fenda sináptica, e da subsensibilização de receptores pós-sinápticos.1
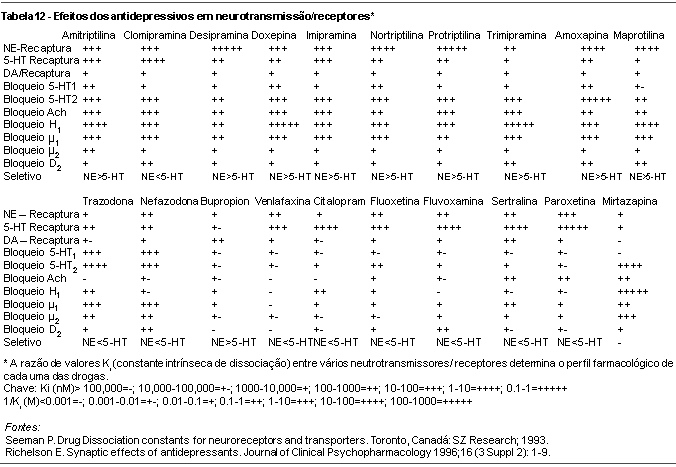
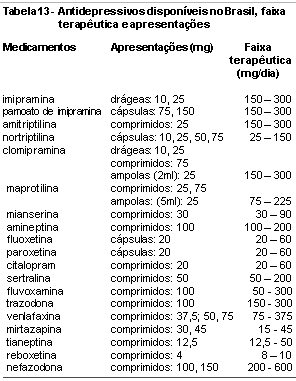
Comparando os novos antidepressivos aos clássicos ADTs e IMAOs, verifica-se um esforço no sentido de aperfeiçoar cada vez mais a ação em sítios receptores determinantes da eficácia clínica, evitando aqueles responsáveis pelos efeitos colaterais. Do amplo espectro de ação dos antidepressivos clássicos passou-se aos ISRSs, melhor tolerados, seguros na superdosagem e de ação praticamente restrita à inibição da recaptação da serotonina. Apesar dos estudos existentes igualarem sua eficácia aos ADTs, permanecem dúvidas em relação à resposta terapêutica em deprimidos graves.23 Diferenças em farmacocinética e potencial de interações medicamentosas tornam o grupo heterogêneo, passível de indicações em diferentes situações clínicas. Os novos antidepressivos buscavam aliar o amplo espectro de ação dos antidepressivos clássicos à tolerabilidade e segurança dos ISRSs. Assim surgiram moclobemida, trazodona, nefazodona, bupropion, reboxetina, mirtazapina e venlafaxina, entre outros. Também são diferentes nos aspectos farmacocinéticos e no potencial de interação com outras substâncias, tornando-os úteis clinicamente em diversos grupos de pacientes. Para vários antidepressivos novos a segurança em gestantes e lactentes ainda não foi estabelecida. Quanto a amplidão do espectro de ação neuroquímico corresponde a um real ganho em termos de eficácia clínica requer ser testado de maneira mais consistente. As tabelas 11,12 e 13 apresentam resumidamente os principais efeitos dos antidepressivos atualmente disponíveis na neurotransmissão, sua ação nos diferentes receptores, bem como suas apresentações e faixas terapêuticas preconizadas.
Correspondência: Ricardo A. Moreno
Rua Tabapuã 500 conj. 4 - São Paulo, SP. CEP: 04533-909, Telefone: +55-11-8225945
2. Supervisora do Grupo de Doenças Afetivas do Instituto de Psiquiatria do Hospital das Clínicas da FMUSP.
3. Pós-graduanda do Departamento de Psiquiatria da FMUSP e colaboradora do Grupo de Estudos de Doenças Afetivas do Instituto de Psiquiatria do Hospital das Clínicas da FMUSP.
- 1. Paykel ES. Handbook of Affective Disorders.2nd ed. New York: The Guilford Press; 1992.
- 2. Stahl SM. Psychopharmacology of Antidepressants. London: Martin Dunitz; 1997.
- 3. Kessel JB , Simpson GM.Tryciclic and Tetracyclic Drugs. In: Kaplan HI , Sadock BJ, editors. Comprehensive Textbook of Psychiatry. 6th ed. Baltimore: Williams e Wilkins; 1995. p. 2096-112
- 4. Moreno DH, Moreno RA. Depressőes resistentes a Tratamento: proposta de abordagem. J Bras Psiquiatria 1993; 42 (Supl.1):415-55.
- 5. Bezchlibnyk-Butler KZ, Jeffries JJ. Clinical handbook of psychotropic drugs. 9th ed. Seattle: Hogrefe & Huber; 1999.
- 6. Himmelhoch JM. Monoamine oxidase inhibitors. In: Kaplan HI, Sadock BJ, editors. Comprehensive textbook of psychiatry. 6th ed. Baltimore: Williams & Wilkins; 1995. p. 2038-56
- 7. Ciraulo DA, Shader RI, Greenblatt DJ, Creelman W. Drug interactions in psychiatry. 2nd ed. Baltimore (Maryland): Williams & Wilkins; 1995.
- 8. Melo M, Moreno RA. Inibidores da monaminooxidase (IMAOs): qual a melhor dieta?. Boletim de Transtornos Afetivos e Alimentares 1998; 3:4-5.
-
9United States Pharmacopeial. Dispensing Information (USP-DI). Drug Information for the Health Care Professional. Massachusetts: World Color Book Services; 1999.
- 10. Moreno RA. A escolha do antidepressivo nos transtornos de humor: papel da nortriptilina. J Bras Psiquiatria 1992; 41 (supl): 33S-35S.
- 11. Moreno RA, Moreno DH. Antidepressivos Tricíclicos. In: Cordás TA, Moreno RA, editores.Condutas em Psiquiatria. 3Ş ediçăo. Săo Paulo: Lemos Editorial; 1999.p. 135-161.
- 12. Goodnick PJ, Goldstein BJ. Selective serotonin reuptake inhibitors in affective disorders I: Basic pharmacology. J Psychopharmacol 1998;12 (3 suppl B): S3-S20.
- 13. Goldstein BJ, Goodnick PJ. SSRIs in the treatment of affective disorders III. Tolerability, safety and pharmacoeconomics. J Clin Psychopharmacol 1998; 12 (3 suppl B): S55-S88.
- 14. Horst WD, Preskorn SH. Mechanism of action and clinical characteristics of three atypical antidepressants: venlafaxine, nefazodone, bupropion. J Affective Disord 1998; 51: 237-54.
- 15. Feighner JP. Mechanism of Action of Antidepressant Medications. J Clin Psychiatry 1999; 60( suppl 4): 4-11.
- 16. Dewan MJ, Anand VS. Evaluating the Tolerability of the Newer Antidepressants. J Nerv Ment Dis 1999; 187: 96-101.
- 17. Kaplan HI, Sadock BJ. Synopsis of psychiatry. 8th ed. Baltimore: Williams & Wilkins; 1998.
- 18. Healy D, Healy H. The clinical pharmacologic profile of reboxetine: does it involve the putative neurobiological substrates of weelbeing? J Affective Disord 1998; 51: 313-22.
- 19. Montgomery SA. Reboxetine: additional benefits to the depressed patient. J Psychopharmacol 1997; 11 (4 suppl): S9-S16.
-
20Press Release, Pharmacia&Upjohn, 1997.
- 21. Mucci M. Reboxetine: a review of antidepressant tolerability. J Psychopharmacol 1997; 11(4 suppl): S33-S38.
- 22. Fawcett J, Barkin RL. Review of the clinical studies on the efficacy, safety and tolerability of mirtazapine for the treatment of patients with major depression. J Affective Disord 1998; 51: 267-86.
- 23. Amsterdam JD. SSRI efficacy in severe and melancholic depression. J Psychopharmacol 1998;12(suppl B):S99-S111.
anexo 1

Datas de Publicação
-
Publicação nesta coleção
06 Jun 2000 -
Data do Fascículo
Maio 1999