SUMMARY
Isolated growth hormone deficiency (IGHD) is the most common pituitary hormone deficiency and, clinically, patients have delayed bone age. High sequence similarity between CYP21A2 gene and CYP21A1P pseudogene poses difficulties for exome sequencing interpretation. A 7.5 year-old boy born to second-degree cousins presented with severe short stature (height SDS −3.7) and bone age of 6 years. Clonidine and combined pituitary stimulation tests revealed GH deficiency. Pituitary MRI was normal. The patient was successfully treated with rGH. Surprisingly, at 10.8 years, his bone age had advanced to 13 years, but physical exam, LH and testosterone levels remained prepubertal. An ACTH stimulation test disclosed a non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency explaining the bone age advancement and, therefore, treatment with cortisone acetate was added. The genetic diagnosis of a homozygous mutation in GHRHR (p.Leu144His), a homozygous CYP21A2 mutation (p.Val282Leu) and CYP21A1P pseudogene duplication was established by Sanger sequencing, MLPA and whole-exome sequencing. We report the unusual clinical presentation of a patient born to consanguineous parents with two recessive endocrine diseases: non-classic congenital adrenal hyperplasia modifying the classical GH deficiency phenotype. We used a method of paired read mapping aided by neighbouring mis-matches to overcome the challenges of exome-sequencing in the presence of a pseudogene.
INTRODUCTION
Isolated growth hormone deficiency (IGHD) is the most common pituitary hormone deficiency; it can be congenital or acquired. Although the most distinctive clinical manifestation is growth failure, there are many other clinical features including a delayed bone age. Amongst the congenital cases, a genetic aetiology can be established in about 10% of patients, with a higher prevalence in familial (34%) compared with sporadic (4%) IGHD. The main genetic causes to date are deleterious mutations in the genes encoding growth hormone (GH), (GH1) or the receptor for GHRH (GHRHR) (11. Alatzoglou KS, Webb EA, Le Tissier P, Dattani MT Isolated growth hormone deficiency (GHD) in childhood and adolescence: recent advances. Endocr Rev. 2014;35(3):376-432.).
Congenital adrenal hyperplasia is a genetically heterogeneous disorder, most frequently caused by recessive, loss of function mutations in the 21-hydroxylase enzyme, encoded by CϒP21A2, and it can have a wide spectrum of clinical manifestations. The non-classic form can be asymptomatic or associated with signs of postnatal androgen excess: rapid growth, advanced bone age, precocious pubarche, menstrual abnormalities, hirsutism, acne, and/or infertility (22. Araújo RS, Mendonca BB, Barbosa AS, Lin CJ, Marcondes JA, Billerbeck AE, et al. Microconversion between CYP21A2 and CY-P21A1P promoter regions causes the nonclassical form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92(10):4028-34.,33. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21(3):245-91.). Non-classic CAH due to 21-hydroxylase deficiency is caused mainly by recombination between CYP21A2 and a nearly identical pseudogene, CYP21A1P (44. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. 2009;10(1):77-82.).
Here we report the unusual presentation of a boy with IGHD and advanced bone age due to GHRHR and non-classic CYP21A2 mutations. To our knowledge, the association of these two conditions has not been reported previously. Furthermore, the pitfalls of Sanger and whole-exome sequencing (WES) to reach the genetic diagnosis are discussed.
CASE REPORT
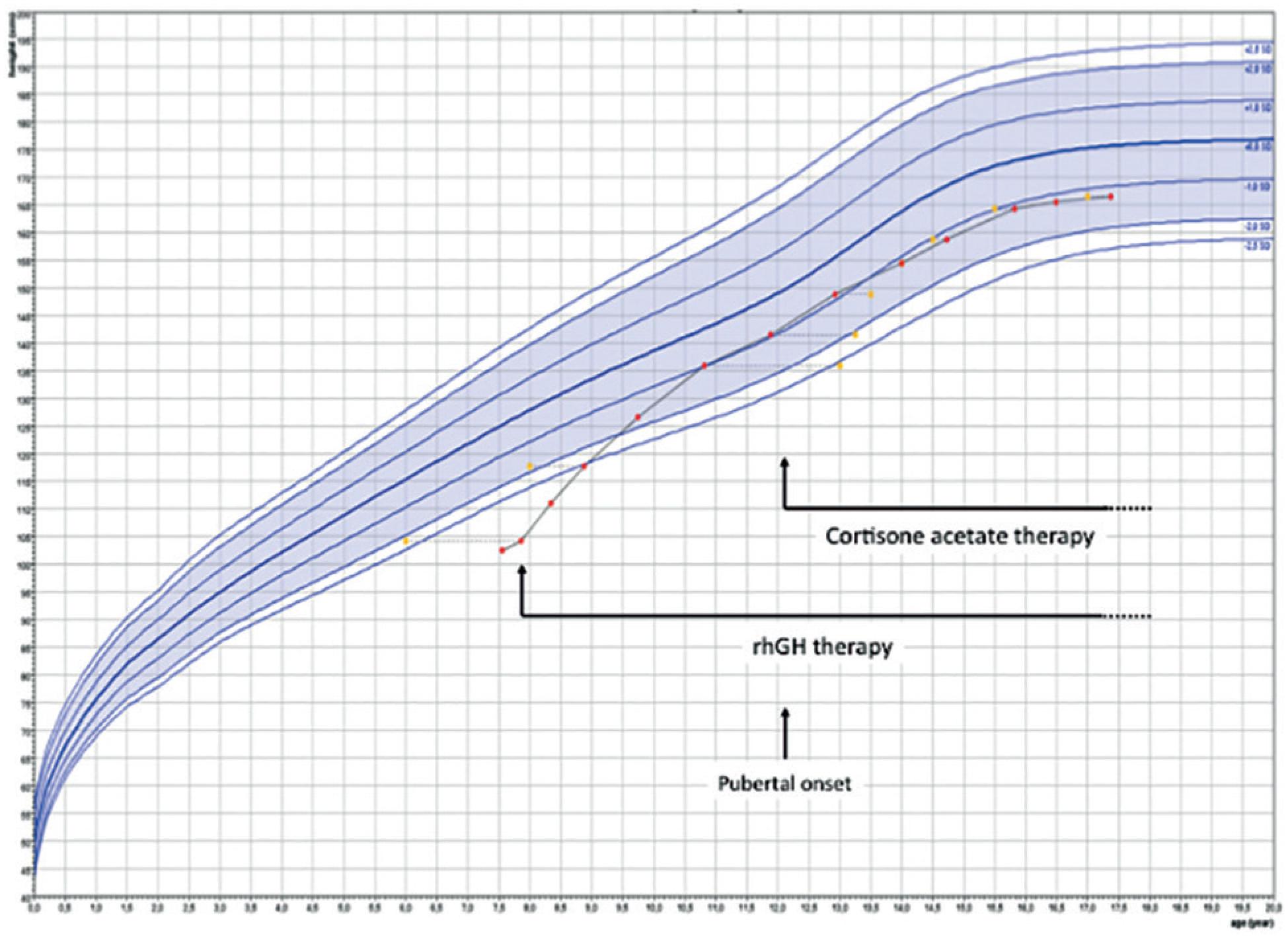
A boy born at term by vaginal delivery (length 50 cm, weight 3,400 g) to second-degree cousins presented at 7.5 years with severe short stature (102.5 cm, SDS-3.7), high-pitched voice, blue sclera and prominent forehead. Genital examination revealed Tanner stage I, absence of pubic hairs, normal penile length (4 cm) and prepubertal testes (length 1.5 cm). Bone age was 6 years according to the Greulich and Pyle method. Basal cortisol (14.8 μg/dL) and FT4 (1.2 ng/dL) were normal. Clonidine stimulation test resulted in a peak GH of 0.6 ng/mL indicating GH deficiency. A combined (insulin, TRH, GnRH) pituitary stimulation tests, performed as part of a research protocol, also showed a peak GH of 0.6 ng/mL and a peak cortisol of 16.1 mcg/dL, initially interpreted as partial ACTH deficiency. Pituitary MRI was normal: anterior lobe 4.6 mm height (normal for age 4.5 ± 0.6 mm) (55. Argyropoulou M, Perignon F, Brunelle F, Brauner R, Rappaport R. Height of normal pituitary gland as a function of age evaluated by magnetic resonance imaging in children. Pediatr Radiol. 1991;21(4):247-9.), normal pituitary stalk and topic posterior pituitary. The patient was successfully treated with rGH (33 mcg/ kg/day) with a first-year growth velocity of 11.7 cm/ year. Surprisingly, at 10.8 years of age he presented with advanced bone age (13 years), (Figure 1), despite absence of signs of puberty and prepubertal serum LH and testosterone levels. Growth velocity at this point was 8.6 cm/year. An ACTH-stimulation test showed respectively, basal and peak levels, cortisol 6.1 and 18.8 mcg/dL, 17 hydroxyprogesterone 9.4 and 52.0 ng/mL and androstenedione 1.2 and 2.0 ng/mL, indicating non-classic 21-hydroxylase deficiency. The spectrum of clinical manifestations in non-classic CAH is wide and there is no perfect genotype-phenotype correlation (66. Moura-Massari VO, Bugano DD, Marcondes JA, Gomes LG, Mendonca BB, Bachega TA. CYP21A2 genotypes do not predict the severity of hyperandrogenic manifestations in the nonclassical form of congenital adrenal hyperplasia. Horm Metab Res. 2013;45(4):301-7). We probably found the advanced bone age prior to pubarche because we were checking it periodically due to IGHD. In common clinical settings, pubarche is usually the presenting sign in boys with non-classic CAH. Cortisone acetate (15 mg/m2/day) was added to his treatment. At 12.2 years of age pubic hair was Tanner stage 2 and testicular length had increased to 2.5 cm indicating onset of central puberty. At 19.5 years, his adult height was 166.5 cm, below his target height of 178.5 cm, possibly due to his early bone age advancement due to CAH (Figure 2).
Advanced bone age (13 years) in a patient with chronological age of 10.8 years with isolated growth hormone deficiency.
Growth curve. Treatment with recombinant human GH (rhGH) and corstisona acetate are shown. Bone age was determined by the Greulich and Pyle criteria. Growth chart was drawn using Growth analyser 3.5 (Ed. Dutch Growth Foundation, PO Box 23068, 3001 KB, Rotterdam, The Netherlands).
GENETIC TESTING AND DISCUSSION
At first, we performed Sanger-sequencing. No mutations in GH1, GHRH, or GHRHR were found (77. Marui S, Trarbach EB, Boguszewski MC, França MM, Jorge AA, Inoue H, et al. GH-releasing hormone receptor gene: a novel splice-disrupting mutation and study of founder effects. Horm Res Paediatr. 2012;78(3):165-72.). Using specific primers for the active CϒP21A2 gene (88. Billerbeck A, Mendonca B, Pinto E, Madureira G, Arnhold I, Bachega T. Three novel mutations in CYP21 gene in Brazilian patients with the classical form of 21-hydroxylase deficiency due to a founder effect. J Clin Endocrinol Metab. 2002;87(9):4314-7), a homozygous c.844G>T, p.Val282Leu mutation (previously known as p.Val281Leu) was found (both parents were heterozygous) (Figure 3A). The p.Val282Leu mutation in CYP21A2 is the most commonly found mutation in patients with non-classic CAH (44. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. 2009;10(1):77-82.) and leads to a mild mutant that retains 2050% of 21-hydroxylase activity (99. Tusie-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265(34):20916-22.).
Panel A: Sanger sequencing of CYP21A2 with specific primers showing the mutation c.844G>T (p.Val282Leu) in homozygous state in the patient and in heterozygous state in the parents. Panel B: The Integrative Genomics Viewer (IGV) showing Alignment without special bioinformatics treatment suggesting heterozygous state of the CYP21A2 c.844G>T (p.Val282Leu) mutation. Panel C: Diagram of the CYP21A2 and CYP21A1P genes. The p.Val282Leu mutation and genomic position is marked as red. The four mis-matches that were used for pair read mapping between CYP21A2 and CYP21A1P sequences are in blue.
In order to establish the unidentified genetic cause of IGHD, WES was performed. Briefly, we aligned sequence reads to the 1000 Genomes Phase 1 reference mapped to GRCh37 using BWA v0.5.9 (1010. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754-60.) and removed duplicate read pairs using PICARD v1.74. We performed realignment, recalibration and variant calling using GATK v3.3 (1111. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-303.) and applied GATK VQSR filter (1212. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491-8.) to remove low-quality variants. Variant annotation was retrieved by using ANNOVAR (1313. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.) revealing a homozygous c.431C>T, p.Leu144His mutation in GHRHR. This mutation is indeed detected in the homozygous state by Sanger sequencing, but it was originally overlooked by the first Sanger sequencing, patient 5 of reference (77. Marui S, Trarbach EB, Boguszewski MC, França MM, Jorge AA, Inoue H, et al. GH-releasing hormone receptor gene: a novel splice-disrupting mutation and study of founder effects. Horm Res Paediatr. 2012;78(3):165-72.). This alteration was not seen when the sequencing data were initially read but after the WES it could be found in the original Sanger sequencing. Here we report the correct GHRHR findings for this patient. This mutation has been previously described in unrelated patients with IGHD from Sergipe/Brazil, Pakistan, Spain and the United States, and has been reported to lead to reduced cAMP production after GHRH stimulation with normal cell- surface localization of the receptor, suggesting a defect in ligand binding (1414. Salvatori R, Fan X, Phillips JA, Espigares-Martin R, Martin De Lara I, Freeman KL, et al. Three new mutations in the gene for the growth hormone (gh)-releasing hormone receptor in familial isolated GH deficiency type IB. J Clin Endocrinol Metab. 2001;86(1):273-9.). To explore a possible founder effect we analyzed the C/T polymorphisms at positions −261 and −235 of the GHRHR promoter, away 7333 and 7359 bp, respectively, from the c.431C>T mutation. Our patient was homozygous C at position −235 and homozygous C at position −261. This haplotype is identical to that of the previously reported patients with p.Leu144His from north-eastern Brazil and Spain but different from the patient from north-eastern United States (1515. Salvatori R, Aguiar-Oliveira MH, Monte LV, Hedges L, Santos NL, Pereira RM, et al. Detection of a recurring mutation in the human growth hormone-releasing hormone receptor gene. Clin Endocrinol (Oxf). 2002;57(1):77-80.). Therefore, the present patient is not related to the previously reported patient from the United States; however, a common ancestor for both families from Brazil and that of Spain, who share the same haplotype, cannot be excluded. Interestingly, another homozygous GHRHR mutation, c.57+1G>A, found in Itabaianinha in the Northeastern Brazilian state of Sergipe, affects the largest kindred of patients with IGHD due to GHRHR mutations reported to date (1616. Salvatori R, Hayashida CY, Aguiar-Oliveira MH, Phillips JA, Souza AH, Gondo RG, et al. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab. 1999;84(3):917-23.).
As we already had the WES done, we decided to analyse CϒP21A2 by this method. At first, interpretation of WES indicated the CYP21A2 p.Val282Leu mutation in heterozygous state (Figure 3B). This was in disagreement with the clinical diagnosis and Sanger sequencing. MLPA, was performed and, revealed CYP21A1P and C4B duplication. To resolve this gene- pseudogene twist in exome-sequencing, we proposed a method of paired read mapping aided by neighbouring four mis-matches using the exome-sequencing data and manually sorted out the real genotype for the variant of interest at CYP21A2 (Figure 3C). For the CYP21A2 mutation, there were 44 paired reads supporting the T allele and 0 supporting the G allele. We concluded that the genotype at CYP21A2 c.844 position is T/T. For the corresponding CYP21A1P mutation, the evidence showed 78 paired reads supporting T and 73 supporting G; thus, it should be G/T at corresponding CYP21A1P position. We believe that these difficulties may happen when analyzing mutations in genes with pseudogenes and highly homologous sequences and this methodology can be useful to overcome this limitation.
Ectopic posterior pituitary lobe and an interrupted stalk on MRI are increasingly being used for the diagnosis of GHD. However, it should be noted that all patients with GHD reported to date with mutations in GH1 and GHRHR (as well as in PROP1) have had a normal stalk and topic posterior lobe, as the present case did (1717 Osorio MG, Marui S, Jorge AA, Latronico AC, Lo LS, Leite CC, et al. Pituitary magnetic resonance imaging and function in patients with growth hormone deficiency with and without mutations in GHRH-R, GH-1, or PROP-1 genes. J Clin Endocrinol Metab. 2002;87(11):5076-84.,1818. Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, et al. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. 2009;94(9):3191-9.).
We conclude that in patients with IGHD and advanced bone age clinicians should search for an additional diagnosis. Patients born to consanguineous parents may have more than one genetic disease leading to unusual phenotypes and treatment outcomes. Whole- exome sequencing was able to establish the genetic cause of IGHD but initially presented difficulties in diagnosing the genotype of CYP21A2/CYP21A1P. This report reveals the strengths and challenges of each sequencing technology and its applications.
-
Funding: this work was supported by the National Council of Technological and Scientific Development - Brazil: grant number CNPq-PQ 305743/2011-2 to BBM; grant number CNPq-PQ 307922/2013-8 to IJPA and grant number CNPq- PQ 304678/2012-0 to AALJ and by São Paulo Research Foundation - Fapesp: grant number 2013/03236-5 to AALJ, and by the National Institutes of Health - United States of America: grant number R01HD030428 to SAC.
REFERENCES
-
1Alatzoglou KS, Webb EA, Le Tissier P, Dattani MT Isolated growth hormone deficiency (GHD) in childhood and adolescence: recent advances. Endocr Rev. 2014;35(3):376-432.
-
2Araújo RS, Mendonca BB, Barbosa AS, Lin CJ, Marcondes JA, Billerbeck AE, et al. Microconversion between CYP21A2 and CY-P21A1P promoter regions causes the nonclassical form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92(10):4028-34.
-
3White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21(3):245-91.
-
4Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. 2009;10(1):77-82.
-
5Argyropoulou M, Perignon F, Brunelle F, Brauner R, Rappaport R. Height of normal pituitary gland as a function of age evaluated by magnetic resonance imaging in children. Pediatr Radiol. 1991;21(4):247-9.
-
6Moura-Massari VO, Bugano DD, Marcondes JA, Gomes LG, Mendonca BB, Bachega TA. CYP21A2 genotypes do not predict the severity of hyperandrogenic manifestations in the nonclassical form of congenital adrenal hyperplasia. Horm Metab Res. 2013;45(4):301-7
-
7Marui S, Trarbach EB, Boguszewski MC, França MM, Jorge AA, Inoue H, et al. GH-releasing hormone receptor gene: a novel splice-disrupting mutation and study of founder effects. Horm Res Paediatr. 2012;78(3):165-72.
-
8Billerbeck A, Mendonca B, Pinto E, Madureira G, Arnhold I, Bachega T. Three novel mutations in CYP21 gene in Brazilian patients with the classical form of 21-hydroxylase deficiency due to a founder effect. J Clin Endocrinol Metab. 2002;87(9):4314-7
-
9Tusie-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265(34):20916-22.
-
10Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754-60.
-
11McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-303.
-
12DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491-8.
-
13Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
-
14Salvatori R, Fan X, Phillips JA, Espigares-Martin R, Martin De Lara I, Freeman KL, et al. Three new mutations in the gene for the growth hormone (gh)-releasing hormone receptor in familial isolated GH deficiency type IB. J Clin Endocrinol Metab. 2001;86(1):273-9.
-
15Salvatori R, Aguiar-Oliveira MH, Monte LV, Hedges L, Santos NL, Pereira RM, et al. Detection of a recurring mutation in the human growth hormone-releasing hormone receptor gene. Clin Endocrinol (Oxf). 2002;57(1):77-80.
-
16Salvatori R, Hayashida CY, Aguiar-Oliveira MH, Phillips JA, Souza AH, Gondo RG, et al. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab. 1999;84(3):917-23.
-
17Osorio MG, Marui S, Jorge AA, Latronico AC, Lo LS, Leite CC, et al. Pituitary magnetic resonance imaging and function in patients with growth hormone deficiency with and without mutations in GHRH-R, GH-1, or PROP-1 genes. J Clin Endocrinol Metab. 2002;87(11):5076-84.
-
18Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, et al. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. 2009;94(9):3191-9.
Publication Dates
-
Publication in this collection
Dec 2017
History
-
Received
11 Feb 2016 -
Accepted
09 May 2017