Abstracts
Chagas' disease is a major health problem in Latin America, where it constitutes one of the leading causes of heart failure. About one fourth of Trypanosoma cruzi-infected individuals develop chronic chagasic cardiomyopathy (CChC), the most severe form of the disease. CChC is histologically characterized by the presence of multifocal inflammatory infiltrates in the heart, composed mainly by mononuclear cells, usually adhered to myocytes and leading to myocytolysis, and frequently by interstitial fibrosis. The pathogenesis of CChC is still unclear, despite intense investigations both in human beings and in animal models of the disease. Although tissue parasitism is rare in the chronic phase of infection, an immune response targeted to persistent parasites or parasite antigens is suggested, by some authors, as the pathogenic mechanism of CChC. Other researchers affirm that the lack of correlation between tissue parasitism and intensity of inflammation suggests, along with the presence of autoreactive immune responses, that CChC results from the action of an autoimmune response. Herein we review reports from the literature and our own data, which together indicate, on one hand, the participation of parasite-specific immune responses and, on the other hand, clearly demonstrate the participation of heart-specific immune responses in the pathogenesis of CChC. Moreover, multiple factors may determine whether an individual in the indeterminate form of the disease will develop CChC. The mechanisms by which T. cruzi breaks immunological tolerance to heart antigens are also discussed.
Chagas' disease; myocarditis; autoimmunity; Trypanosoma cruzi; delayed-type hypersensitivity
A doença de Chagas constitui um grave problema de saúde pública na América Latina, onde é uma das principais causas de problemas cardíacos. A cardiopatia chagásica crônica (CChC), forma mais grave da doença, manifesta-se em cerca de 25% dos indivíduos infectados pelo Trypanosoma cruzi, e é caracterizada, a nível histopatológico, pela presença de infiltrados multifocais de células mononucleares, freqüentemente aderidas a miócitos e induzindo miocitólise, sendo também bastante comum o achado de fibrose intersticial. Embora a doença tenha sido intensamente estudada, tanto em seres humanos como em modelos animais, o mecanismo de patogênese da CChC ainda é discutido. Apesar da escassez de parasitas na fase crônica da doença, uma possível resposta imune contra os parasitas persistentes seria, para alguns, o mecanismo de patogênese da CChC. Por outro lado, a falta de correlação entre o parasitismo tecidual e intensidade de inflamação sugere, juntamente com a presença de respostas autoreativas, que a CChC resulte da ação de um mecanismo autoimune. Neste artigo, revisamos dados da literatura e dados obtidos em nossos laboratórios, que sugerem a participação de mecanismos efetores ativados em resposta a antígenos parasitários e demonstram um papel de respostas autoreativas contra antígenos cardíacos na patogênese da CChC. Múltiplos fatores devem contribuir para a progressão da forma indeterminada para a CChC. Os possíveis mecanismos ativados pela infecção por T. cruzi que levam à quebra de tolerância imunológica a antígenos cardíacos são discutidos.
Doença de Chagas; miocardite; auto imunidade; Trypanosoma cruzi; reação de hipersensibilidade tardia
The pathogenesis of Chagas' disease: when autoimmune and parasite-specific immune responses meet* * Invited paper Correspondence to: Dr. Ricardo Ribeiro dos Santos E-mail: rrsantos@cpqgm.fiocruz.br
MILENA B. P. SOARES1, LAIN PONTES-DE-CARVALHO2, 3
and RICARDO RIBEIRO-DOS-SANTOS1, 3
1Laboratório de Imunofarmacologia and 2Laboratório de Imunologia Celular e Molecular
Centro de Pesquisas Gonçalo Moniz/FIOCRUZ, Salvador, BA, Brazil
3Escola Bahiana de Medicina e Saúde Pública, Salvador, BA, Brazil
Manuscript received on September 19, 2001; accepted for publication on September 26, 2001;
presented by ANTONIO C. CAMPOS-DE-CARVALHO
ABSTRACT
Chagas' disease is a major health problem in Latin America, where it constitutes one of the leading causes of heart failure. About one fourth of Trypanosoma cruzi-infected individuals develop chronic chagasic cardiomyopathy (CChC), the most severe form of the disease. CChC is histologically characterized by the presence of multifocal inflammatory infiltrates in the heart, composed mainly by mononuclear cells, usually adhered to myocytes and leading to myocytolysis, and frequently by interstitial fibrosis. The pathogenesis of CChC is still unclear, despite intense investigations both in human beings and in animal models of the disease. Although tissue parasitism is rare in the chronic phase of infection, an immune response targeted to persistent parasites or parasite antigens is suggested, by some authors, as the pathogenic mechanism of CChC. Other researchers affirm that the lack of correlation between tissue parasitism and intensity of inflammation suggests, along with the presence of autoreactive immune responses, that CChC results from the action of an autoimmune response. Herein we review reports from the literature and our own data, which together indicate, on one hand, the participation of parasite-specific immune responses and, on the other hand, clearly demonstrate the participation of heart-specific immune responses in the pathogenesis of CChC. Moreover, multiple factors may determine whether an individual in the indeterminate form of the disease will develop CChC. The mechanisms by which T. cruzi breaks immunological tolerance to heart antigens are also discussed.
Key words: Chagas' disease, myocarditis, autoimmunity, Trypanosoma cruzi, delayed-type hypersensitivity.
INTRODUCTION
Infection with Trypanosoma cruzi causes American trypanosomiasis or Chagas' disease, which progresses in three consecutive phases. An acute phase, characterized by intense parasitism and blood parasitemia occurs after parasite transmission. This is followed by an asymptomatic or indeterminate phase, in which no clinical symptoms are observed. Most of the individuals will remain in this phase of the infection throughout the rest of their lives. About 30% of them, however, develop a chronic phase, after time periods ranging from a few months to decades (Dias and Coura 1997). The majority of cases of chronic disease consists of a progressively debilitating chronic cardiomyopathy, for which there is no current cure. One of the leading causes of heart failure in several Latin American countries, the chronic chagasic cardiomyopathy (CChC) is characterized by an intense myocarditis, which may lead to heart enlargement, and frequently by arrythmia, causing, in concert, heart malfunction and death (Köberle 1968).
The pathological basis of CChC has been a matter of intense debate in the last decades. That it is immunologically mediated has been clearly shown by experiments in athymic nude mice, in which infection by T. cruzi causes strikingly high tissue parasitism without heart inflammation (Soares et al. 2001a and Figure 1). The specificity of the pathogenic immune response, however, is the source of the controversy. One main hypothesis proposes that the pathogenic inflammatory response is exclusively directed to T. cruzi antigens at sites of parasite persistence (Tarleton 2001, Higuchi 1997). The second main hypothesis is that CChC is an autoimmune disease triggered in some individuals by T. cruzi infection (Cunha-Neto and Kalil 1995, Leon and Engman 2001). Here, points of view against and in favor of these two hypotheses will be presented, together with evidence that both T. cruzi- and heart-specific responses may be important in the pathology of CChC.
T. CRUZI INFECTION AND THE MURINE MODEL
T. cruzi is a hemoflagellate protozoan parasite capable of infecting different cell types in the mammalian host. It has, frequently, a preference to invade muscle cells, including heart muscle cells. In the acute phase of infection, parasites are easily found replicating in different tissues and organs, as well as circulating in the blood. As adaptive immune responses are stimulated, the high tissue parasitism is controlled, but never eradicated. Host and pathogen will then live in equilibrium, similar to other parasitic infections, such as leishmaniases and toxoplasmosis. This equilibrium may be broken, for instance, in a state of immunosuppression, when patent parasitism may reappear (Sartori et al. 1998, Galhardo et al. 1999).
The evaluation of the factors responsible for the development or control of pathogenic responses has been hindered by the high variability of the parasite: different T. cruzi strains display distinct biological behaviors (Andrade and Magalhães 1996). It has been reported that a single individual was simultaneously infected by different strains of T. cruzi, each showing a preference for specific tissues (Vago et al. 2000). To overcome the difficulties of carrying out experiments with human material, several researchers have chosen to investigate the pathological mechanisms of CChC in a mouse model of infection. Decades of investigations using this experimental model have taught us that both host and parasite genetic backgrounds contribute to disease development. Thus, certain mouse strains will develop CChC after infection with some strains of T. cruzi, but not with others (Andrade 1990, Andrade et al. 1985). A model that closely resembles the human disease has been studied in our laboratories. This model uses the Colombian strain of T. cruzi, a strain isolated from a patient with severe CChC (Federici et al. 1964). Infection of BALB/c mice with Colombian strain trypomastigotes, using an infective inoculum of 100 parasites, similar to what occurs in natural transmission (Brener et al. 2000), reproduces the three phases of infection observed in human beings. In the acute phase of infection, high parasitemia and intense myocarditis is observed, followed by a period of nearly complete regression of heart inflammation. However, four to five months after the onset of parasitemia, an intense and progressive inflammatory response in heart tissue is found.
PARASITE PERSISTENCE AND INFLAMMATION
The first aspect to be analyzed is the scarce parasitism found in the chronic phase of infection. Several studies have demonstrated that T. cruzi parasites or antigens can be found, although rarely, in individuals with chronic infection (Barbosa Jr and Andrade 1984, Jones et al. 1993, Palomino et al. 2000). However, the analyses of human heart fragments have failed to show a correlation between intensity of inflammation and of parasitism, even with the use of highly sensitive techniques, such as PCR or immunohistochemistry (Palomino et al. 2000, Olivares-Villagomez et al. 1998). In fact, this remarkable feature of the disease was immediately pointed out 90 years ago by Vianna (1911), the excellent pathologist working in collaboration with Carlos Chagas. Vianna described that the intensity of inflammatory response observed in hearts of individuals with chagasic cardiopathy was not proportional to the degree of tissue parasitism. More importantly, in CChC patients, rare parasites are also found in other organs free of significant inflammation, whereas in the myocardium, they are found associated with chronic inflammatory response and fibrosis (Barbosa Jr and Andrade 1984, Mortara et al. 1999). This has also been observed in the mouse model of CChC (Buckner et al. 1999). If parasite persistence were the only factor driving the pathological response, one would expect a clear association between parasitism and inflammation, which would be manifested in all parasitized organs. The fact that this is not so is a clear evidence that the chronic inflammation is not merely caused by a T. cruzi-specific immune response.
DELAYED-TYPE HYPERSENSITIVITY AND CChC
The inflammatory reaction observed in CChC has a delayed-type hypersensitivity (DTH)-like aspect, with focal inflammation composed mainly of mononuclear cells (Andrade 1983). DTH reactions to T. cruzi antigens were shown in vivo in human beings (Mayer and Pifano 1941, Tschudi et al. 1968) and in the mouse model using crude T. cruzi extracts (Gonzales-Cappa et al. 1968), as well as purified T. cruzi antigens, such as the immunodominant SAPA antigen (Nasser et al. 1997) and cruzipain (Laderach et al. 1996). The DTH responses are mediated by CD4+ T cells, as adoptive transfer of this cell population purified from T. cruzi-infected mice to naïve mice also transferred the ability to mount a local DTH reaction (Hontebeyrie-Joskowicz et al. 1987). As pointed out above, the lack of association between tissue parasitism and inflammation suggest that the pathogenic response in CChC is not only directed against T. cruzi antigens but also to self antigens. However, anti- T. cruzi responses can also induce lesions and contribute to CChC. In the acute phase of infection, destruction of intracardiac parasympathetic fibers occur, which may in part be responsible to some of the complications observed in the chronic phase of infection (Ribeiro-dos-Santos et al. 1976). This neuronal destruction is probably mediated by inflammatory reactions promoted by anti- T. cruzi responses targeted to T. cruzi antigens adsorbed on the neuronal cell surface (Ribeiro-dos-Santos and Hudson 1980a,b).
The DTH aspect of inflammatory reactions in CChC suggests that Th1 cells participate in the pathogenesis of this disease. In fact, an association between intensity of CChC and production of high levels of IFN-g in human infection has been shown (Bahia-Oliveira et al. 1998). To investigate the role of IFN-g T-cell responses in the pathogenesis of experimental CChC, we studied the course of T. cruzi infection in IL-4-deficient BALB/c mice, in which Th1 responses are accentuated. Infection of these mice with Colombian strain T. cruzi caused higher acute phase parasitemia and tissue parasitism than the infection of IL-4 +/+ control mice (Soares et al. 2001a). These findings correlated with the production of IFN-g, since IL-4 -/- mice had higher levels of this cytokine than IL-4 +/+ mice. Interestingly, despite the lower number of parasites, IL-4 -/- mice had an early and more intense myocarditis than IL-4 +/+ mice (Figure 2). These results indicate a dual role of IFN-g in T. cruzi infection, where this cytokine has a protective role in parasitism control, but, on the other hand, acts as a critical mediator of CChC. Moreover, they reinforce the lack of association between intensity of inflammation and degree of tissue parasitism in CChC. This lack of association is also observed when T. cruzi-infected young and aged mice are compared. Despite of having lower parasitemia and mortality early in the infection, aged mice have more intense myocarditis in the chronic phase of infection than young mice (Cardillo et al. 1993). This phenomenon may also be associated with a decreased Th2 response, as aged mice have impaired Th2-dependent immune responses (Smith et al. 2001).
AUTOIMMUNE RESPONSES IN CChC
Proving a disease to be of autoimmune aetiology is not an easy task. To classify a disease as autoimmune, one needs to show not only the presence of an immune response against self antigens, but also clinical or experimental evidence of the primary role of this response in the pathogenesis of tissue lesions.
Immune responses against self antigens in human and experimental Chagas' disease were demonstrated by several research groups. Antibodies against a number of antigens, including antigens expressed in cardiac (McCornick and Rowland 1989, Cunha-Neto et al. 1995) and nervous (Ribeiro-dos-Santos et al. 1979, Van Voorhis and Eisen 1989) tissues, and UsnRNPs (Bach-Elias et al. 1998), among others, have been detected during T. cruzi infection (reviewed in Kierszenbaum 1999). However, autoantibodies are commonly found after infection with different pathogens, without any implications regarding an autoimmune pathology (Argov et al. 1989, Daniel-Ribeiro and Zanini, 2000).
As for autoantibodies, there is a growing number of reports indicating a pathogenic role of IgG antibodies, present in the sera of chagasic patients, against muscarinic and adrenergic receptors of cardiomyocytes (Borda et al. 1984, Sterin-Borda et al. 1991). The production of autoantibodies against beta1 and beta2-adrenoreceptors correlated with primary electrical cardiac abnormalities (Chiale et al. 1995). It was also demonstrated that sera from chronic chagasic individuals interfere with electric and mechanical activities of embryonic myocardial cells in vitro (Costa et al. 2000, Kaplan et al. 1997) and affect cardiac electrogenesis and conduction when perfused in the rabbit heart (de Oliveira et al. 1997). These alterations in heart-cell function seem to result from the binding of antibodies to  -adrenergic and M2-cholinergic receptors on the myocardial cell surface (Costa et al. 2000, de Oliveira et al. 1997, Kaplan et al. 1997). Thus, by stimulating the production of autoantibodies against certain self-antigens, T. cruzi infection may cause disturbances in heart cell functions, which may lead to complications associated with CChC.
-adrenergic and M2-cholinergic receptors on the myocardial cell surface (Costa et al. 2000, de Oliveira et al. 1997, Kaplan et al. 1997). Thus, by stimulating the production of autoantibodies against certain self-antigens, T. cruzi infection may cause disturbances in heart cell functions, which may lead to complications associated with CChC.
Autoreactive T lymphocytes specific to heart or nerve tissue antigens were also found in chagasic mice and patients (Rizzo et al. 1989, Cunha-Neto et al. 1996, Hontebeyrie-Joskowicz et al. 1987). A first indication of a pathogenic role of self-reactive T-cell responses in CChC was obtained by our group using a heterotopic heart transplant model (Ribeiro-dos-Santos et al. 1992). In this report, newborn syngeneic hearts transplanted in the ears of T. cruzi-infected mice were rejected, while the control hearts transplanted in ears of normal mice engrafted and started beating after seven to ten days. The rejection of normal, syngeneic heart transplant was mediated by CD4+ T cells, as the transfer of this T cell population purified from spleens of chagasic mice, but not of CD8+ T cells, rendered normal recipients capable of rejecting syngeneic transplants. A few years later, however, Tarleton et al. (1997) published a report in which they describe that, in a different infection model (using Brazil and Sylvio strains of T. cruzi to infect C57Bl6 and C3H mice), rejection of syngeneic heart transplants did not occur in chronic chagasic mice. Graft rejection was only observed when the transplant was performed during the acute phase of infection (and parasites could be found in the heart graft), or when T. cruzi antigen was injected into the transplanted tissue. These observations lead the authors to the conclusion that an anti- T. cruzi response was responsible for the destruction of the heart transplant. Thus, in some mouse models, parasite-specific immune responses seem to be the only cause of CChC (Tarleton et al. 1997), whereas this does not seem to be the case in other mouse models (Ribeiro-dos-Santos et al. 1992, Buckner et al. 1999) and in human beings (Barbosa Jr and Andrade 1984, Mortara et al. 1999).
To further demonstrate the role of autoreactive T cells in the pathogenesis of experimental CChC, we generated a CD4+ T-cell line from the spleen of a chronic chagasic mouse (Ribeiro-dos-Santos et al. 2001). This T-cell line was raised by repeated in vitro stimulation with syngeneic heart extract. After eight months of culture, this T-cell line strongly reacted with heart extracts from different sources (syngeneic, allogeneic and xenogeneic). Interestingly, this T-cell line also reacted with T. cruzi antigen, although the culture was maintained free of parasites by treatment with amphotericin B. More importantly, this anti-heart T-cell line caused the destruction of myoblasts in vitro as well as syngeneic heart transplant rejection when injected into the transplanted heart. It proliferated in the presence of T. cruzi and heart antigens, and presented a Th1 cytokine profile upon stimulation with these antigens. More importantly, the adoptive transfer of this T-cell line induced intense myocarditis in athymic mice (nu/nu), which is extremely sensitive to T. cruzi infection, in the complete absence of T. cruzi.
AUTOIMMUNE MYOCARDITIS
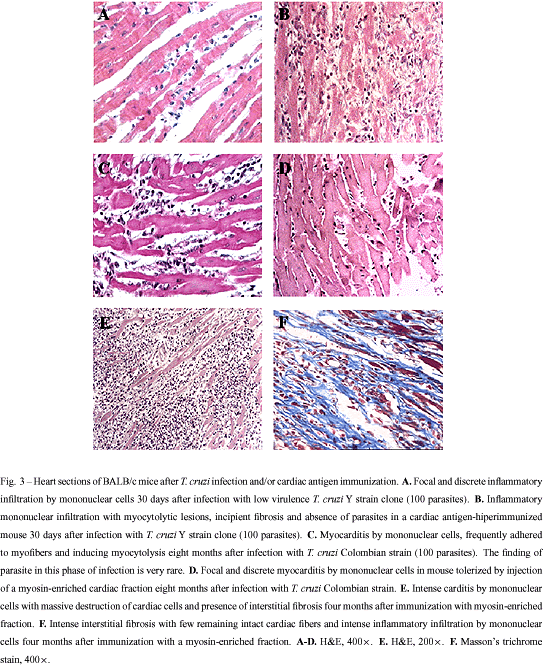
The inflammatory reaction found in CChC is similar to that of autoimmune myocarditis induced by immunization with cardiac myosin, characterized by a multifocal inflammatory infiltrate composed by mononuclear cells and interstitial fibrosis (Figure 3E and F). This similarity lead us and others to compare the immune responses of mice infected with T. cruzi and/or immunized with heart antigens. Leon et al. (2001) demonstrated that sera from A/J mice (highly susceptible to autoimmune myocarditis) contain antibodies which strongly recognized heart antigens, including myosin, in the acute phase of T. cruzi infection. Additionally, T. cruzi-infected A/J mice developed DTH response against cardiac myosin. Based on these observations, the authors concluded that an autoimmune response is triggered by T. cruzi early in the infection. Due to the high virulence of T. cruzi for A mice, the evaluation of hearts from these mice in the chronic phase of infection is difficult. However, A mice treated with benznidazole during the acute phase of infection survive and progress to the chronic phase of infection (authors' unpublished observation). Using this approach, we found that A mice have intense myocarditis after three months of infection with the Colombian-strain T. cruzi, whereas BALB/c mice have nearly absent inflammation at this time after infection. This result shows that A mice are more susceptible than BALB/c mice both for induction of experimental autoimmune myocarditis and for development of CChC, suggesting a common basis for both diseases. Moreover, hyperimmunization (eight injections) of BALB/c mice with low doses of heart antigen does not induce autoimmune myocarditis. However, two months after a challenge with Y strain T. cruzi (100 parasites), these mice had intense myocarditis, whereas infected-control mice have nearly normal hearts (Soares et al. 2001b; Figure 3 A and B). Hyperimmunized BALB/c IL-4 -/- mice, on the other hand, developed mild, but clearcut myocarditis, which was also aggravated after T. cruzi infection. One explanation for these findings is that T. cruzi infection breaks the tolerance to heart antigens, which is kept under control by mechanisms probably dependent on IL-4.
WHAT ARE THE MECHANISMS BY WHICH T. CRUZI BREAKS IMMUNOLOGICAL TOLERANCE?
The main proposed mechanisms by which a microorganism could trigger an autoimmune disease are the sharing of epitopes (molecular mimicry) between the pathogen and the host, and the inflammation and tissue damage caused by the infection, leading to release self antigens, recruitment of inflammatory cells and/or production of immunomodulators and expression of surface molecules, which could, in concert, trigger or sustain an autoimmune response (Davies 1997, Fairweather et al. 2001). One of the better characterized example of molecular mimicry between T. cruzi and a heart antigen was demonstrated by Cunha-Neto and co-workers. These investigators showed the presence of antibodies in chagasic patients which reacted both with cardiac myosin and B13, a T. cruzi antigen (Cunha-Neto et al. 1995). More recently, T-cell clones cross-reacting with cardiac myosin and B13 were also obtained from hearts of CChC patients (Cunha-Neto et al. 1996). Most of these T-cell clones are CD4+ and secrete high levels of IFN-g (Abel et al. 2001). However, there is no demonstration of a role of the response against B13 in the pathogenesis of CChC, neither in human beings, nor in experimental models. In fact, it has been recently suggested that there is no evidence supporting a role for molecular mimicry in the pathogenesis of autoimmune diseases triggered by microorganisms, in general (Benoist and Mathis 2001).
T. cruzi could also act triggering or enhancing an autoreactive response kept under control by suppressive mechanisms. First, by its ability to infect cardiac fibers, it causes intense damage, antigen release and inflammation in the acute phase of infection. Second, T. cruzi may affect the properties of professional antigen-presenting cells, such as macrophages and dendritic cells. In this regard, it was demonstrated that T. cruzi molecules can modulate the production of pro-inflammatory cytokines by macrophages (Almeida et al. 2000), and affect the maturation of dendritic cells (Van Overtvelt et al. 1999). Third, the immune response against T. cruzi antigens could also influence the induction of self-reactive responses. In experimental T. cruzi infection, high levels of IFN-g are produced during the acute phase of the disease (Hoft et al. 1993, Zhang and Tarleton 1996). This production may result from the parasite intensely stimulating macrophages to produce IL-12 and IL-18, two potent IFN-g inducing factors (Frosch et al. 1996, Meyer zum Büschenfelde et al. 1997, Camargo et al. 1997). Additionally, the immunodominant T. cruzi trans-sialidase superfamily antigens induce Th1-type responses both in human beings and in mice (Millar and Kahn 2000, Ribeirão et al. 2000). Thus, the persistence of parasites could sustain the release of self antigens, the presence of small inflammatory foci in the heart and the production of inflammatory cytokines, which may, in concert, potentiate a self-reactive response. The possibility of re-infection with T. cruzi of individuals living in endemic areas may also influence the development of CChC, as new loads of parasites may induce release of autoantigens and soluble mediators. In fact, it has been reported that the frequency of infected individuals developing CChC has decreased in areas where parasite transmission has been controlled (Dias and Coura 1997).
CAN WE BLOCK THE PROGRESSION OF CChC?
Understanding the mechanisms of pathology in CChC may help the development of a therapy for prevention or intervention on the disease progress. Based on the results obtained with heart antigen immunization, we used the mouse model to test protocols of immunomodulation of CChC by induction of tolerance to heart antigens. Tolerance was induced by co-administration of anti-CD4 monoclonal antibodies and a myosin-enriched fraction, prepared from syngeneic hearts, before infection with Colombian strain T. cruzi. Hearts from tolerized mice had no or very discrete myocarditis eight months after infection, whereas hearts from non-tolerized mice had intense inflammation (Pontes-de-Carvalho et al. 2001; Figure 3C and D). This result clearly demonstrates a role for an autoimmune component in CChC, which could be prevented or inhibited by induction of tolerance to heart antigens.
CONCLUDING REMARKS
Perhaps the most intriguing feature of CChC is the fact that most T. cruzi-infected individuals will survive throughout their lives without any manifestations of the disease. This may be due to many factors, such as the parasite strain, the host genetic background, the number of re-infections, or even the success of the treatment. Concerning this last point, it is noteworthy the lack of market availability of new anti- T. cruzi drugs over the past 20 years. It is clear that, although an autoimmune response to heart antigens participate in the pathogenesis of CChC, the persistence of parasite or parasite antigens (Andrade et al. 2000) may be required for triggering or sustaining it. The development of heart failure or disfunction may also depend on multiple events occurring in different phases of infection, such as neuronal destruction and activity of autoantibodies to cardiac beta-adrenergic and muscarinic membrane receptors, in addition to myocardial inflammation.
ACKNOWLEDGMENTS
The authors' works were supported by CNPq and UNDP/Word Bank/WHO-TDR.
RESUMO
A doença de Chagas constitui um grave problema de saúde pública na América Latina, onde é uma das principais causas de problemas cardíacos. A cardiopatia chagásica crônica (CChC), forma mais grave da doença, manifesta-se em cerca de 25% dos indivíduos infectados pelo Trypanosoma cruzi, e é caracterizada, a nível histopatológico, pela presença de infiltrados multifocais de células mononucleares, freqüentemente aderidas a miócitos e induzindo miocitólise, sendo também bastante comum o achado de fibrose intersticial. Embora a doença tenha sido intensamente estudada, tanto em seres humanos como em modelos animais, o mecanismo de patogênese da CChC ainda é discutido. Apesar da escassez de parasitas na fase crônica da doença, uma possível resposta imune contra os parasitas persistentes seria, para alguns, o mecanismo de patogênese da CChC. Por outro lado, a falta de correlação entre o parasitismo tecidual e intensidade de inflamação sugere, juntamente com a presença de respostas autoreativas, que a CChC resulte da ação de um mecanismo autoimune. Neste artigo, revisamos dados da literatura e dados obtidos em nossos laboratórios, que sugerem a participação de mecanismos efetores ativados em resposta a antígenos parasitários e demonstram um papel de respostas autoreativas contra antígenos cardíacos na patogênese da CChC. Múltiplos fatores devem contribuir para a progressão da forma indeterminada para a CChC. Os possíveis mecanismos ativados pela infecção por T. cruzi que levam à quebra de tolerância imunológica a antígenos cardíacos são discutidos.
Palavras-chave: Doença de Chagas, miocardite, auto imunidade, Trypanosoma cruzi, reação de hipersensibilidade tardia.
PONTES-DE-CARVALHO L, SANTANA CC, SOARES MBP, OLIVEIRA GGS, CONRADO-DOS-SANTOS WL, CUNHA-NETO E AND RIBEIRO-DOS-SANTOS R. Murine chronic Chagasic cardiomyopathy is an autoallergic disease that can be prevented by the reinforcement of immunological self-tolerance, (submitted).
SOARES MBP, LIMA RS, SANTANA CC, BELLINTANI MC, PONTES-DE-CARVALHO LC AND RIBEIRO-DOS-SANTOS R. 2001b. Repeated immunization with syngeneic heart antigens sensitizes mice to Trypanosoma cruzi-induced myocarditis.
- ABEL LC, RIZZO LV, IANNI B, ALBUQUERQUE F, BACAL F, CARRARA D, BOCCHI EA, TEIXEIRA HC, MADY C, KALIL J, AND CUNHA-NETO E. 2001. Chronic Chagas' Disease Cardiomyopathy Patients Display an Increased IFN-gamma Response to Trypanosoma cruzi Infection. J Autoimmun 17: 99-107.
- ALMEIDA IC, CAMARGO MM, PROCOPIO DO, SILVA LS, MEHLERT A, TRAVASSOS LR, GAZZINELLI RT AND FERGUSON MA. 2000. Highly purified glycosylphosphatidylinositols from Trypanosoma cruzi are potent proinflammatory agents. EMBO J 19: 1476-1485.
- ANDRADE SG. 1990. Influence of Trypanosoma cruzi strain on the pathogenesis of chronic myocardiopathy in mice. Mem Inst Oswaldo Cruz 85: 17-27.
- ANDRADE ZA. 1983. Mechanisms of myocardial damage in Trypanosoma cruzi infection. Ciba Found Symp 99: 214-233.
- ANDRADE SG AND MAGALHĂES JB. 1996. Biodemes and zymodemes of Trypanosoma cruzi strains: correlations with clinical data and experimental pathology. Rev Soc Bras Med Trop 30: 27-35.
- ANDRADE SG, PIMENTEL AR, DE SOUZA MM AND ANDRADE ZA. 2000. Interstitial dendritic cells of the heart harbor Trypanosoma cruzi antigens in experimentally infected dogs: importance for the pathogenesis of chagasic myocarditis. Am J Trop Med Hyg 63: 64-70.
- ANDRADE V, BARRAL-NETTO M, ANDRADE SG AND MAGALHAES JB. 1985. Immunological aspects of infection of 6 inbred strains of mice by 3 different strains of Trypanosoma cruzi Mem Inst Oswaldo Cruz 80: 203-211.
- ARGOV S, JAFFE CL, KRUPP M, SLOR H AND SHOENFELD Y. 1989. Autoantibody production by patients infected with Leishmania. Clin Exp Immunol 76: 190-197.
- BACH-ELIAS M, BAHIA D, TEIXEIRA DC AND CICARELLI RM. 1998. Presence of autoantibodies against small nuclear ribonucleoprotein epitopes in Chagas' patients' sera. Parasitol Res 84: 796-799.
- BAHIA-OLIVEIRA LM, GOMES JA, ROCHA MO, MOREIRA MC, LEMOS EM, LUZ ZM, PEREIRA ME, COFFMAN RL, DIAS JC, CANCADO JR, GAZZINELLI G AND CORREA-OLIVEIRA R. 1998. IFN-gamma in human Chagas' disease: protection or pathology? Braz J Med Biol Res 31: 127-131.
- BARBOSA JR AA AND ANDRADE ZA. 1984. Identificaçăo do Trypanosoma cruzi nos tecidos extracardíacos de portadores de miocardite crônica chagásica. Ver Soc Bras Med Trop 17: 123-126.
- BENOIST C AND MATHIS D. 2001. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol 2: 797-801.
- BORDA EP, COSSIO P, VEGA MV, ARANA R AND STERIN-BORDA L. 1984. A circulating IgG in Chagas disease which binds to ß-adrenoreceptors of myocardium and modulates their activity. Clin Exp Immunol 57: 679-686.
- BRENER Z, ANDRADE ZA AND BARRAL-NETTO M. 2000. Trypanosoma cruzi e doença de Chagas, 2nd ed., Rio de Janeiro: Guanabara Koogan, 431p.
- BUCKNER FS, WILSON AJ AND VAN VOORHIS WC. 1999. Detection of live Trypanosoma cruzi in tissues of infected mice by using histochemical stain for beta-galactosidase. Infect Immun 67: 403-409.
- CAMARGO MM, ALMEIDA IC, PEREIRA MES, FERGUSON MAJ, TRAVASSOS LR AND GAZZINELLI RT. 1997. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes initiate the synthesis of proinflammatory cytokines by macrophages. J Immunol 158: 5890-5901.
- CARDILLO F, FALCAO RP, ROSSI MA AND MENGEL J. 1993. An age-related gamma delta T cell suppressor activity correlates with the outcome of autoimmunity in experimental Trypanosoma cruzi infection. Eur J Immunol 23: 2597-2605.
- CHIALE PA, ROSENBAUM MB, ELIZARI MV, HJALMARSON A, MAGNUSSON Y, WALLUKAT G AND HOEBEKE J. 1995. High prevalence of antibodies against beta1 and beta2-adrenoreceptors in patients with primary electrical cardiac abnormalities. J Am Coll Cardiol 26: 864-869.
- COSTA PC, FORTES FS, MACHADO AB, ALMEIDA NA, OLIVARES EL, CABRAL PR, PEDROSA RC, GOLDENBERG RC, CAMPOS-DE-CARVALHO AC AND MASUDA MO. 2000. Sera from chronic chagasic patients depress cardiac electrogenesis and conduction. Braz J Med Biol Res 33: 439-446.
- CUNHA-NETO E AND KALIL J. 1995. Autoimmunity in Chagas' heart disease. Rev Paul Med 113: 757-766.
- CUNHA-NETO E, DURANTI M, GRUBER A, ZINGALES B, DE MESSIAS I, STOLF N, BELLOTTI G, PATARROYO ME, PILLEGGI F AND KALIL J. 1995. Autoimmunity in Chagas disease cardiopathy: biological relevance of a cardiac myosin-specific epitope crossreactive to an immunodominant Trypanosoma cruzi antigen. Proc Natl Acad Sci USA 92: 3541-3545.
- CUNHA-NETO E, COELHO V, GUILHERME L, FIORELLI A, STOLF N, KALIL J. 1996. Autoimmunity in Chagas' disease. Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas' cardiomyopathy patient. J Clin Invest 98: 1709-1712.
- DANIEL-RIBEIRO CT AND ZANINI G. 2000. Autoimmunity and malaria: what are they doing together? Acta Trop 76: 205-221.
- DAVIES JM. 1997. Molecular mimicry: can epitope mimicry induce autoimmune disease? Immunol Cell Biol Apr 75(2): 113-26.
- DE OLIVEIRA SF, PEDROSA RC, NASCIMENTO JH, CAMPOS DE CARVALHO AC AND MASUDA MO. 1997. Sera from chronic chagasic patients with complex cardiac arrhythmias depress electrogenesis and conduction in isolated rabbit hearts. Circulation 96: 2031-2037.
- DIAS JCP AND COURA J. 1997. Clínica e terapęutica da doença de Chagas: uma abordagem prática para o clínico geral, Rio de Janeiro: FIOCRUZ, 486p.
- FAIRWEATHER D, KAYA Z, SHELLAM GR, LAWSON CM AND ROSE NR. 2001. From infection to autoimmunity. J Autoimmun 16: 175-186.
- FEDERICI EE, ABELMANN WB AND NEVA FA. 1964. Chronic and progressive myocarditis in C3H mice infected with Trypanosoma cruzi Am J Trop Med Hyg 13: 272-280.
- FROSCH S, SWANTJE K AND FLEISHER B. 1996. Trypanosoma cruzi is a potent inducer of interleukin-12 production in macrophages. Med Microbiol Immunol 185: 189-193.
- GALHARDO MCG, MARTINS IA, HASSLOCHER-MORENO A, XAVIER SS, COELHO JMC, VASCONCELOS ACV AND RIBEIRO-DOS-SANTOS R. 1999. Reactivation of Trypanosoma cruzi infection in a patient with acquired immunodeficiency syndrome. Rev Soc Bras Med Trop 32: 291-294.
- GONZALES-CAPPA SM, SCHMUNIS GA, TRAVERSA OC, YANOVSKY JF AND PARODI AS. 1968. Complement fixation test, skin test and experimental immunization with antigens of Trypanosoma cruzi prepared under pressure. Am J Trop Med Hyg 17: 709-715.
- HIGUCHI ML. 1997. Chronic chagasic cardiopathy: the product of a turbulent host-parasite relationship. Rev Inst Med Trop Săo Paulo 39: 53-60.
- HOFT DF, LYNCH RG AND KIRCHHOFF LV. 1993. Kinetic analysis of antigen-specific immune responses in resistant and susceptible mice during infection with Trypanosoma cruzi J Immunol 151: 7038-7047.
- HONTEBEYRIE-JOSKOWICZ M, SAID G, MILON G, MARCHAL G AND EISEN H. 1987. L3T4+ T cells able to mediate parasite-specific delayed-type hypersensitivity play a role in the pathology of experimental Chagas' disease. Eur J Immunol 17: 1027-1033.
- JONES EM, COLLEY DG, TOSTES S, LOPES ER, VNENCAK-JONES CL AND MCCURLEY TL. 1993. Amplification of a Trypanosoma cruzi DNA sequence from inflammatory lesions in human chagasic cardiomyopathy. Am J Trop Med Hyg 48: 348-357.
- KAPLAN D, FERRARI I, BERGAMI PL, MAHLER E, LEVITUS G, CHIALE P, HOEBEKE J, VAN REGENMORTEL MH AND LEVIN MJ. 1997. Antibodies to ribosomal P proteins of Trypanosoma cruzi in Chagas disease possess functional autoreactivity with heart tissue and differ from anti-P autoantibodies in lupus. Proc Natl Acad Sci USA 94: 10301-10306.
- KIERSZENBAUM F. 1999. Chagas' disease and the autoimmunity hypothesis. Clin Microbiol Rev 12: 210-223.
- KÖBERLE F. 1968. Chagas' disease and Chagas' syndromes: the pathology of American Trypanosomiasis. Adv Parasitol 6: 63-116.
- LADERACH D, CERBAN F, MOTRAN C, VOTTERO DE CIMA E AND GEA S. 1996. Trypanosoma cruzi: the major cysteinyl proteinase (cruzipain) is a relevant immunogen of parasite acidic antigens (FIII). Int J Parasitol 26: 1249-1254.
- LEON JS AND ENGMAN DM. 2001. Autoimmunity in Chagas heart disease. Int J Parasitol 31: 555-561.
- LEON JS, GODSEL LM, WANG K AND ENGMAN DM. 2001. Cardiac myosin autoimmunity in acute chagas' heart disease. Infect Immun 69: 5643-5649.
- MAYER M AND PIFANO CF. 1941. O diagnóstico da moléstia de Chagas por intradermo reaçăo com cultivo de Schizotrypanum cruzi Brasil Med 55: 317-319.
- MCCORNICK TS AND ROWLAND EC. 1989. Trypanosoma cruzi: cross-reactive anti-heart autoantibodies produced during infection in mice. Exp Parasitol 69: 393-401.
- MEYER ZUM BÜSCHENFELDE C, CRAMER S, TRUMPFHELLER C, FLEISHER B AND FRO S. 1997. Trypanosoma cruzi induces strong IL-12 and IL-18 gene expression in vivo: correlation with interferon-gamma (IFN-g) production. Clin Exp Immunol 110: 378-385.
- MILLAR AE AND KAHN SJ. 2000. The SA85-1.1 protein of the Trypanosoma cruzi trans-sialidase superfamily is a dominant T-cell antigen. Infect Immun 68: 3574-3580.
- MORTARA RA, DA SILVA S, PATRICIO FR, HIGUCHI ML, LOPES ER, GABBAI AA, CARNEVALE P, ROCHA A, FERREIRA MS, SOUZA MM, DE FRANCO MF, TURCATO G JR AND FERRAZ NETO BH. 1999. Imaging Trypanosoma cruzi within tissues from chagasic patients using confocal microscopy with monoclonal antibodies. Parasitol Res 85:800-808.
- NASSER JR, GOMEZ LE, SANCHEZ D, GUERIN M AND BASOMBRIO MA. 1997. Immunogenicity of the recombinant SAPA protein of Trypanosoma cruzi for mice. J Parasitol 83: 76-81.
- OLIVARES-VILLAGOMEZ D, MCCURLEY TL, VNENCAK-JONES CL, CORREA-OLIVEIRA R, COLLEY DG AND CARTER CE. 1998. Polymerase chain reaction amplification of three different Trypanosoma cruzi DNA sequences from human chagasic cardiac tissue. Am J Trop Med Hyg 59: 563-570.
- PALOMINO SA, AIELLO VD AND HIGUCHI ML. 2000. Systematic mapping of hearts from chronic chagasic patients: the association between the occurrence of histopathological lesions and Trypanosoma cruzi antigens. Ann Trop Med Parasitol 94: 571-579.
- RIBEIRĂO M, PEREIRA-CHIOCCOLA VL, RENIA L, AUGUSTO FRAGATA FILHO A, SCHENKMAN S AND RODRIGUES MM. 2000. Chagasic patients develop type I immune response to Trypanosoma cruzi trans-sialidase. Parasite Immunol 22: 49-53.
- RIBEIRO-DOS-SANTOS R AND HUDSON L. 1980a. Trypanosoma cruzi: binding of parasite antigens to mammalian cell membranes. Paras Immunol 2: 1-10.
- RIBEIRO-DOS-SANTOS R AND HUDSON L. 1980b. Trypanosoma cruzi: immunological consequences of parasite modification of host cells. Clin Exp Immunol 40: 36-41.
- RIBEIRO-DOS-SANTOS R, RAMOS DE OLIVEIRA JC AND KOBERLE F. 1976. Aspectos imunopatológicos da destruiçăo neuronal na moléstia de Chagas. Rev Goiana de Med 22: 235-243.
- RIBEIRO-DOS-SANTOS R, MARQUEZ JO, VON GAL FURTADO CC, RAMOS DE OLIVEIRA JC, MARTINS AR AND KOBERLE F. 1979. Antibodies against neurons in chronic Chagas' disease. Tropenmed Parasitol 30: 19-23.
- RIBEIRO-DOS-SANTOS R, ROSSI MA, LAUS JL, SILVA JS, SAVINO W AND MENGEL J. 1992. Anti-CD4 abrogates rejection and reestablishes long-term tolerance to syngeneic newborn hearts grafted in mice chronically infected with Trypanosoma cruzi J Exp Med 175: 29-39.
- RIBEIRO-DOS-SANTOS R, MENGEL JO, POSTOL E, SOARES RA, FERREIRA-FERNANDEZ E, SOARES MB AND PONTES-DE-CARVALHO LC. 2001. A heart-specific CD4+ T-cell line obtained from a chronic chagasic mouse induces carditis in heart-immunized mice and rejection of normal heart transplants in the absence of Trypanosoma cruzi Parasite Immunol 23: 93-101.
- RIZZO LV, CUNHA-NETO E AND TEIXEIRA AR. 1989. Autoimmunity in Chagas' disease: specific inhibition of reactivity of CD4+ T cells against myosin in mice chronically infected with Trypanosoma cruzi Infect Immun 57: 2640-2644.
- SARTORI AM, LOPES MH, BENVENUTI LA, CARAMELLI B, DI PIETRO A, NUNES EV, RAMIREZ LP AND SHIKANAI-YASUDA MA. 1998. Reactivation of Chagas' disease in a human immunodeficiency virus-infected patient leading to severe heart disease with a late positive direct microscopic examination of the blood. Am J Trop Med Hyg 59: 784-786.
- SMITH P, DUNNE DW AND FALLON PG. 2001. Defective in vivo induction of functional type 2 cytokine responses in aged mice. Eur J Immunol 31: 1495-1502.
- SOARES MBP, SILVA-MOTA KN, LIMA RS, BELLINTANI MC, PONTES-DE-CARVALHO L AND RIBEIRO-DOS-SANTOS R. 2001. Modulation of chagasic cardiomyopathy by interleukin-4: dissociation between inflammation and tissue parasitism. Am J Pathol 159: 703-709.
- STERIN-BORDA L, GORELIK G AND BORDA ES. 1991. Chagasic IgG binding with cardiac muscarinic cholinergic receptors modifies cholinergic-mediated cellular transmembrane signals. Clin Immunol Immunopathol 61: 387-397.
- TARLETON RL. 2001. Parasite persistence in the aetiology of Chagas disease. Int J Parasitol 31: 550-554.
- TARLETON RL, ZHANG L AND DOWNS MO. 1997. "Autoimmune rejection" of neonatal heart transplants in experimental Chagas disease is a parasite-specific response to infected host tissue. Proc Natl Acad Sci USA 94: 3932-3937.
- TSCHUDI EI, ANZIANO DF AND DALMASSO AP. 1968. Lymphocyte transformation in Chagas disease. Infect Immun 6: 905-908.
- VAGO AR, ANDRADE LO, LEITE AA, D'AVILA REIS D, MACEDO AM, ADAD SJ, TOSTES S JR, MOREIRA MC, FILHO GB AND PENA SD. 2000. Genetic characterization of Trypanosoma cruzi directly from tissues of patients with chronic Chagas disease: differential distribution of genetic types into diverse organs. Am J Pathol 156: 1805-1809.
- VAN OVERTVELT L, VANDERHEYDE N, VERHASSELT V, ISMAILI J, DE VOS L, GOLDMAN M, WILLEMS F AND VRAY B. 1999. Trypanosoma cruzi infects human dendritic cells and prevents their maturation: inhibition of cytokines, HLA-DR, and costimulatory molecules. Infect Immun 67: 4033-4040.
- VAN VOORHIS WC AND EISEN H. 1989. Fl-160. A surface antigen of Trypanosoma cruzi that mimics mammalian nervous tissues. J Exp Med 169: 641-652.
- VIANNA GO. 1911. Contribuiçăo para o estudo da anatomia patológica da "Moléstia de Carlos Chagas" (Esquizotripanoze humana ou tireoidite parasitária). Mem Inst Oswaldo Cruz 3: 276-294.
- ZHANG L AND TARLETON RL. 1996. Characterization of cytokine production in murine Trypanosoma cruzi infection by in situ immunocytochemistry: lack of association between susceptibility and type 2 cytokine production. Eur J Immunol 26: 102-109.
Publication Dates
-
Publication in this collection
12 Dec 2001 -
Date of issue
Dec 2001
History
-
Accepted
26 Sept 2001 -
Received
19 Sept 2001