Abstracts
OBJECTIVE: To extend the molecular analysis of the IVS30+1G>T intronic thyroglobulin (TG) mutation, and to report the eleven year follow-up of the affected patients. METHOSD: Two siblings with severe congenital hypothyroidism with fetal and neonatal goiter, harboring the IVS30+1G>T mutation were included. Nodular and non-nodular thyroid tissue specimens were collected. Specific thyroid genes expression was evaluated by real-timePCR and by immunohistochemistry. RESULTS: In non-nodular tissue specific thyroid genes mRNA were reduced when compared to normal thyroid sample. In the nodule, TPO and NIS expression was very low. Microscopic examinations showed very large follicular-lumina and swollen vesicles of endoplasmatic-reticulum. Strong cytoplasmatic and low follicular-lumen TG immunostaining were detected. Intracellular NIS, membrane TPO and TSHR immunostaining had higher positivity in non-nodular sample. Both patients had a long-term adequate developmental outcome, besides one patient have been lately-treated. CONCLUSIONS: IVS30+1G>T mutation not only lead to very enlarge endoplasmatic-reticulum, but also to alterations of specific thyroid genes expression. The clinical evolution of patients harboring these mutations strengthen the concept of the influence of environment, like iodine nutrition, to determine the final phenotypic appearance.
Thyroglobulin; Congenital hypothyroidism; Gene mutations; Molecular diagnosis; Molecular analysis
OBJETIVO: Aprofundar a análise molecular da mutação intrônica IVS30+1G>T do gene tireoglobulina (TG) e relatar a clínica de pacientes portadores da mutação, acompanhados por 11 anos. MÉTODOS: Foram estudados dois irmãos com hipotireoidismo congênito grave com bócio fetal e bócio neonatal, portadores da mutação IVS30+1G>T. Foram coletadas amostras de tecido nodular e não-nodular. Avaliou-se a expressão de genes específicos da tireóide por PCR em tempo real e imunohistoquímica. RESULTADOS: A expressão de genes específicos da tireóide foi menor no tecido não-nodular que no tecido normal controle. Expressões de TPO e NIS foram extremamente baixas no tecido nodular. Verificou-se lúmen folicular aumentado com grandes vesículas de retículo endoplasmático, e detectou-se forte marcação de TG no citoplasma e fraca no lúmen folicular. No tecido não-nodular observou-se forte positividade de NIS intracelular e, TPO e TSHR na membrana plasmática. O acompanhamento em longo prazo dos pacientes mostrou adequado desenvolvimento, apesar de um deles ter recebido tratamento tardio. CONCLUSÕES: A mutação IVS30+1G>T não só promove alterações no retículo endoplasmático, como alterações na expressão de genes específicos da tireóide. A evolução clínica destes pacientes reforça o conceito da influência do meio ambiente, como o aporte nutricional de iodo, no fenótipo final.
Tireoglobulina; Hipotireoidismo congênito; Mutação gênica; Diagnóstico molecular; Estudo molecular
CLINICAL CASE REPORT
A molecular analysis and long-term follow-up of two siblings with severe congenital hypothyroidism carrying the IVS30+1G>T intronic thyroglobulin mutation
Análise molecular e acompanhamento a longo prazo de dois irmãos com hipotireoidismo congênito portadores da mutação intrônica IVS30+1G>T no gene da tireoglobulina
Ileana G. S. RubioI; Ana Luiza GalraoI; Viviane PardoI; Meyer KnobelI; Roberta F. PossatoI; Rosalinda R. Y. CamargoI; Marcelo A. FerreiraII; Cristina T. KanamuraIII; Simone A. GomesIV; Geraldo Medeiros-NetoI
IThyroid Unit (LIM-25), Division of Endocrinology, University of São Paulo Medical School, São Paulo, SP, Brazil
IICell Biology (LIM59), Division of Pathology, University of São Paulo Medical School, São Paulo, SP, Brazil
IIIAdolfo Lutz Institute, São Paulo Public Health Service, São Paulo, SP, Brazil
IVDivision of Endocrinology, Federal University of Sergipe Medical School, Aracajú, SE, Brazil
Correspondence to Correspondence to: Ileana G S Rubio Thyroid Unit - LIM 25, University of São Paulo Medical School Av. Dr. Arnaldo, 455 4A 01246-903 São Paulo SP E-mail: ilearubi@usp.br
ABSTRACT
OBJECTIVE: To extend the molecular analysis of the IVS30+1G>T intronic thyroglobulin (TG) mutation, and to report the eleven year follow-up of the affected patients.
METHOSD: Two siblings with severe congenital hypothyroidism with fetal and neonatal goiter, harboring the IVS30+1G>T mutation were included. Nodular and non-nodular thyroid tissue specimens were collected. Specific thyroid genes expression was evaluated by real-timePCR and by immunohistochemistry.
RESULTS: In non-nodular tissue specific thyroid genes mRNA were reduced when compared to normal thyroid sample. In the nodule, TPO and NIS expression was very low. Microscopic examinations showed very large follicular-lumina and swollen vesicles of endoplasmatic-reticulum. Strong cytoplasmatic and low follicular-lumen TG immunostaining were detected. Intracellular NIS, membrane TPO and TSHR immunostaining had higher positivity in non-nodular sample. Both patients had a long-term adequate developmental outcome, besides one patient have been lately-treated.
CONCLUSIONS: IVS30+1G>T mutation not only lead to very enlarge endoplasmatic-reticulum, but also to alterations of specific thyroid genes expression. The clinical evolution of patients harboring these mutations strengthen the concept of the influence of environment, like iodine nutrition, to determine the final phenotypic appearance.
Keywords: Thyroglobulin; Congenital hypothyroidism; Gene mutations; Molecular diagnosis; Molecular analysis
RESUMO
OBJETIVO: Aprofundar a análise molecular da mutação intrônica IVS30+1G>T do gene tireoglobulina (TG) e relatar a clínica de pacientes portadores da mutação, acompanhados por 11 anos.
MÉTODOS: Foram estudados dois irmãos com hipotireoidismo congênito grave com bócio fetal e bócio neonatal, portadores da mutação IVS30+1G>T. Foram coletadas amostras de tecido nodular e não-nodular. Avaliou-se a expressão de genes específicos da tireóide por PCR em tempo real e imunohistoquímica.
RESULTADOS: A expressão de genes específicos da tireóide foi menor no tecido não-nodular que no tecido normal controle. Expressões de TPO e NIS foram extremamente baixas no tecido nodular. Verificou-se lúmen folicular aumentado com grandes vesículas de retículo endoplasmático, e detectou-se forte marcação de TG no citoplasma e fraca no lúmen folicular. No tecido não-nodular observou-se forte positividade de NIS intracelular e, TPO e TSHR na membrana plasmática. O acompanhamento em longo prazo dos pacientes mostrou adequado desenvolvimento, apesar de um deles ter recebido tratamento tardio.
CONCLUSÕES: A mutação IVS30+1G>T não só promove alterações no retículo endoplasmático, como alterações na expressão de genes específicos da tireóide. A evolução clínica destes pacientes reforça o conceito da influência do meio ambiente, como o aporte nutricional de iodo, no fenótipo final.
Descritores: Tireoglobulina; Hipotireoidismo congênito; Mutação gênica; Diagnóstico molecular; Estudo molecular.
INTRODUCTION
Congenital hypothyroidism (CH) is the most common endocrine disease in infancy, with a frequency, approximately, of 1/3000 live births (1). In 85% of patients, the disorder is associated with thyroid development (dysgenesis), and 15%, harbor inborn errors in thyroid hormone synthesis (dyshormonogenesis) (2). Dyshormonogenesis is transmitted as a classical autosomal recessive mendelian trait and the clinical spectrum of the resulting phenotypes ranges from mild to severe goitrous hypothyroidism (3). Thyroglobulin (TG) is a large glycoprotein synthesized by the thyroid gland, and functions as a matrix for thyroid hormone synthesis (4). Thirty-eight inactivating mutations have been identified, characterized in the human TG gene and associated to congenital goiter and hypothyroidism (5). We have previously identified the intronic IVS30+1G>T mutation in two Brazilian families with a complex history of fetal goiter and congenital goiter, born to consanguineous parents (6-8). In this report we have extended our initial molecular and immunlogic studies of two affected goitrous siblings with defective TG synthesis and we describe the eleven year follow-up of the two siblings harboring this mutation.
PATIENTS
We study two siblings with congenital hypothyroidism due to a defective thyroglobulin synthesis and secretion. They were born in Aracajú, coastal city of the northeast of Brazil, to a consanguineous parents (second degree cousins).
Patient 1 (AJM) was born in 1990 before the era of mandatory national neonatal screening for congenital hypothyroidism in Brazil. At birth he presented neonatal goiter, without other typical signal of congenital hypothyroidism. He was referred to an endocrinologic appointment after birth but further investigations were not performed. Unfortunately early medical records were not available. His mother informed that during the first years of live the patient was very calm and she suspected of a delay in development. Congenital hypothyroidism was finally diagnosed at 3 years of age. The child had a good compliance to daily thyroxin treatment. Clinical data and thyroid function tests are shown in Table 1. Bone age was retarded (2yrs 8m) for his chronological age of 5yrs 9m old. At puberty, bone age was similar to chronological age (Table 1). Echographic studies of thyroid gland indicated an enlarged gland at 6yrs old, and erroneously diagnosed as "chronic thyroiditis" (hypoechogeinity). Anti-TPO antibodies were persistently negative. At 12 yrs of age intellectual and somatic development was considered as normal. Puberty (Tanner V) was completed at 14a 5m. During the first years of elementary school he had poor school performance. As an young adult however, he is finishing the regular high school program and is considering to apply for college. During a recent medical examination he had an athletic appearance (heith 1.76m), with a very good verbal communication, talked enthusiastically about his future perspectives, and gave the impression of a normal 17 yrs old adolescence (Figure 1). At that age the volume of the gland was 26.1mL (goiter). Thyroidectomy was indicated due to the presence of two solid nodules (2.7 X 1.5 X 2.0 cm and 1.3 X 1.0 X 1.0 cm), respectively in the right and left lobes. Pathological diagnosis was suggested of dyshormonogenetic goiter. Both nodules were benign adenomas.
Patient 2 the younger sister of Patient 1 was born in 1997. She had a large fetal goiter (12.7mL) revealed by US of her mother at 26 weeks of gestation. Fetal hypothyroidisms was confirmed by cordocentesis (TSH 61.3mU/L; TG 1.3mg/L) (9). After 4 weeks of a single intra-amniotic injection of 400µg L-thyroxine, a marked reduction of the goiter volume was confirmed by ultrasound (from 12.3mL to 4.8mL). She received 12.0ug LT4/kg weight. Clinical data and thyroid function tests are shown in Table 2. When she was 7 years old marked hypoechogeinity of the thyroid gland was diagnosed by ultrasonographic examination, with negative anti-TPO antibodies. She had a normal somatic and intellectual development. Bone age was according with chronological age. Menarche was at 11yrs 4m of age followed by regular cycles (Figure 1).
Absence of synthesis and secretion of TG was confirmed in both siblings by lack of serum TG increment 24 and 48 hours after stimulation of 0.1mg intramuscular injection of recombinant human TSH (rhTSH). Molecular studies of the thyroglobulin gene mutations identified the intronic mutation IVS30+1G>T in both patients (8). Both siblings had elevated urine iodine excretion (respectively, 539 ug/L, and 492ug/L) confirming a relatively high content in the diet. Both parents are euthyroid without history of thyroid disease, and harbored the heterozygous form of the intronic mutation.
METHODS
Thyroid functions tests
Serum total T4 (TT4), serum total T3 (TT3), serum TSH and serum TG levels were determined by electrochemiluminescence immunoassays (Roche Corporation, IN, USA).
Tissue samples
Nodular and non-nodular thyroid tissue specimens were collected from Patient 1 that was submitted to total thyroidectomy. One fragment of the specimen was immediately frozen in liquid nitrogen. Others were kept in formalin for immunohistochemistry and in 2.5% glutaraldehyde for electron microscopy. We also used RNA sample from a normal human thyroid tissue (control tisuue).
Gene expression quantification by real-time PCR
Total RNA was isolated using Trizol LS (Gibco BRL, Life Technologies, Gaithersburg, MD) and cDNA was synthesized with Super Script III RNA-H reverse tanscriptase (Invitrogen, Carlsbad, EUA). We quantified gene expression of TG, Sodium-iodine symporter (NIS), thyroperoxidase (TPO), TSH receptor (TSHR), pendrin (PDS), thyroid transcription factor 1 (TTF1) and paired box transcription factor 8 (PAX-8) using Absolute QPCR SYBR® Green Mix (Abgene, Surrey, United Kingdom) in Rotor-Gene 3000 equipment (Corbett Research, Mortlake, Australia). We also quantified the expression of GAPDH as internal control. PFAFFL (10) method was used to calculate gene expression, reported as relative arbitrary units (AU). We used the expression in normal control tissue as calibrator sample in the PFAFFL formula, therefore expression value of all the genes in normal control was 1AU. The intron spanning primers were describe in Table 3.
Immunohistochemistry analysis
Paraffin-embedded tissue samples were stained by immunoperoxidase (11) with primary NIS antibody (FP5A, Mayo Clinic, Rochester, MN), TSH receptor, TPO and TG antibodies (DakoCytomation, Dako, Glostrup, Denmark). Amplification step was performed with Dako EnVision System, Peroxidase Kit (Dako, Glostrup, Denmark). An immunostaining score was given according to the percentage of follicular cells with protein positive staining as follows: 0 (0%), 1+ (low, 1%-20%), 2+ (moderate, 21%-49%) and 3+ (high, ≥ 50%).
This work was approved by the Ethical Committee of the Hospital das Clínicas, University of São Paulo Medical School, and was conducted according to the Helsinki Declaration. Informed consent was obtained from the parents of the patients.
RESULTS
To determine whether TG and other important thyroid genes were expressed properly in the goitrous tissue of the affected patient, we performed mRNA quantification and immunohistochemical analysis.
Gene expression quantification
We quantified mRNA concentrations of TG, NIS, TPO, TSHR, PDS, TTF1 and PAX-8 genes in nodular and non-nodular thyroid samples from Patient 1 and from a normal control thyroid tissue. Messenger RNA of all genes was detected in all tissue samples (Table 4). In non-nodular tissue TG mRNA was reduced 37% as compared to control thyroid tissue. Similar results were observed for transcription factors PAX-8 (42%) and TTF-1 (33%), and for the TSHR (58%). NIS and TPO expression was slightly reduced when compared to control tissue (9% and 15% of reduction, respectively) and the lowest level was of PDS gene (78% of reduction).
When we compared nodular and non-nodular tissues TG, TSHR, TPO and NIS expression was reduced (Table 4, Figure 2). Even more, TPO and NIS expression was about 150 and 230 times lower, respectively. On the other hand, higher expression of PDS (2-fold), and similar expression of PAX-8 and TTF1 genes were observed in nodular as compared to non-nodular tissues (Table 4, Figure 2).
Thyroid histological, electron microscopy and immunohistochemical analysis
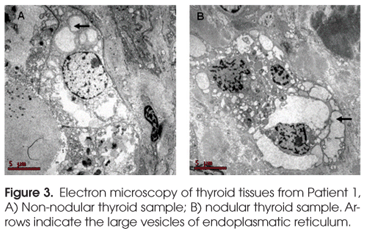
Light microscopic examination of nodular and non-nodular thyroid tissue stained with hematoxilin and eosin demonstrated that the follicle lumina were enlarged and devoid of colloid. Electron microscopy identified cytoplasmatic swollen vesicles of endoplasmatic reticulum (ER) in both samples (Figure 3), in contrast to normally more flattened tubular ER (12).
We performed immunohistochemical detection of TG, NIS, TSHR and TPO proteins. We detected strong (3+) cytoplasmatic TG immunostaining in non-nodular and nodular sample (Table 4). By contrast, very low TG protein was present in the follicular lumen (Figure 3) as opposed to normal thyroid tissue without TG defect (12).
On the other hand, non-nodular sample NIS, TPO and TSHR proteins had higher (+3) positivity when compared with nodular (+1) (Table 4) (Figure 4).
In nodular and non-nodular samples NIS protein localization was intracellular, TPO protein was detected in the apical membrane and TSHR protein had membrane localization (Figure 4).
DISCUSSION
Defective thyroglobulin synthesis usually results in goitrous congenital hypothyroidism. In the present study we have extended the molecular analysis of the IVS30+ 1G>T TG gene mutation and the clinical case of two siblings with congenital hypothyroidism due to thyroglobulin synthesis defect. They were born from consanguineous parents. The analysis of the complete coding sequence of TG gene and intro/exon borders identified the intronic homozygous IVS30+1G>T mutation in both patients (8). The parents harbored the form of this mutations in heterozygous state.
Cases of thyroid carcinoma developing from dysormonogenic goiters have been reported (13,14). Moreover, high incidence of thyroid cancer was associated with long-standing goiters with thyroglobulin mutations (15). Therefore, Patient 1 with the presence of two solid thyroid nodules underwent total thyroidectomy. The pathologic diagnosis of both nodules was benign adenoma.
Intronic mutations of the TG gene with the functional consequence of skipping of an entire exon are not rare in congenital hypothyroid patients (16). The IVS30+1G>T mutation is caused by guanine to thymine transversion at position +1 in the donor splice site of intron 30. This mutations were previously identified by our group in other two siblings from a not related family from the Northeast of Brazil (6). It was also confirmed the compound heterozygous constellation IVS30+1G>T/A2215D in the two first degree cousins of the siblings patients of the present study (8). This intronic mutation promotes aberrant splicing and loss of 138 nucleotides of the TG mRNA, removing the entire exon 30 (6,7). Elimination of this exon does not affect the reading frame of the mRNA and potentially codifies a shortened polypeptide. The deletion is localized in the TG type III repeat domain, causing the loss of 1- putative N-linked glycosylation site (5,17). The loss of 46 aminoacids can modify the tertiary and quaternary structure of the protein. However, silico studies of the mutant protein are not possible because the crystallographic structure of complete TG molecule is not available.
Electron microscopy confirmed the presence of distended endoplasmatic reticulum (ER) both in nodular and non-nodular thyroid samples from patient 1. These results are a consequence of the mutant TG protein retention inside the ER (12). Properly folded not mutant TG dimmers migrates from ER to the Golgi apparatus where glycosilation occurs (18). On the other hand, unfolded mutated protein activated the mechanism of quality control of the ER, mediated by a massive induction of specific ER molecular chaperones including the hsp90 homolog, GRP94, and the hsp70 homolog, BiP, reducing the export of the protein to the colloid and causing thyroid ER storage disease (12). Immunohistochemical analysis confirmed the defective TG traffic both in the non-nodular tissue as well as in the nodule. In both tissues there was a marked decrease of reaction product in the follicular lumina and concomitant accumulation of intracellular staining. The undetectable levels of TG after rhTSH (8) lead us to speculate that an acute stimulation with TSH may not be enough to pass through the ER blockade, whilst that the few molecules that reach the colloid are immediately hydrolyzed after internalization into the thyroid cell.
We were able to detect the mRNA expression of the specific thyroid genes: TG, TPO, NIS, TSHR, TTF1, PAX-8 and PDS in the Patient 1 thyroid tissues, albeit the levels were reduced when compared with normal thyroid tissue. These data may be related to the functional state of the gland. The mRNA data of TG, TPO, NIS and TSHR were in agreement with immunohistochemistry evaluation of protein expression.
In nodule sample the expression levels of the various genes varied widely. TG, TSHR expression was reduced as compare to non-nodular. Furthermore, the highest reduction were those of NIS and TPO expression. NIS expression reduction without decrease of TPO expression levels in benign adenomas have been previously described (19-20). Indeed, in the present study, the expression of NIS and TPO were 150 and 230-fold lower, respectively, in the nodule when compared with non-nodular or normal thyroid tissue. These are two of the key proteins known to regulate iodide uptake and intrathyroid metabolism (20). Consequently, these findings may be related to defects in both the iodine-trapping ability and the iodination process in the nodule. Both physiologic functions were less pronounced in the non-nodular tissue.
Our results indicated that PAX-8, TTF1 and PDS gene expression were still preserved in both nodular and non-nodular samples, however the levels were reduced as compared to normal sample. Previous report have shown that PDS gene and its product (pendrin) expression levels appeared to be similar in most hypofunctioning adenomas, whereas more than a 3-fold decrease was observed in other samples (21,22).
Transcription factors TTF1 an PAX-8 are involved in thyroid development and in the regulation of the expression of specific thyroid genes (TPO, TG, NIS, TSHR, PDS) (23-29) Altogether, our findings of similar and significant expression of PAX-8 and TTF1 in nodular and non-nodular tissue with very low expression of NIS and TPO exclusively in the nodule, suggest that different mechanisms are controlling the expressions of these gene in these tissues.
Our immunostaining analysis indicated membrane localization of TSHR and TPO, and the intracellular localization of NIS proteins. Previous reports have shown predominantly intracellular localization of NIS protein in thyroid tumors (30-32).
Combined, the results of this study and the previously reported molecular analysis (6,7) have proved that IVS30+1G>T TG mutation promotes a severe thyroid hormone synthesis defect with fetal or neonatal goitrous CH (elevated fetal or neonatal TSH and low TG values) (8). On the clinical side, as expected, the three years post birth of Patient 1 without L-thyroxine therapy possibly caused a mild neurological consequences. This was confirmed by the retarded bone age and early intellectual delay. Whilst after a clinical long-term follow-up (eleven years) this patient appears to be a normal adolescent with an actual normal height at 17 years old (1.76m) and presumably good school performance, due to an effective LT4 treatment. We have reasons to believe that elevated iodine nutrition would eventually determine this final phenotypic appearance (8). In the presence of a high iodine supply, the thyroid gland would be able to overcome the severe genetic disease and induce generation of some thyroid hormone, preventing more serious neurological damage (33,34). Urinary iodine excretion in the affected siblings were elevated, indicating that they still are in a relatively high nutritional iodine environment. On the other hand, patient 2 who have received prenatal treatment of fetal hypothyroidism and daily LT4 replacement after birth, have no symptoms of CH during the clinical follow-up.
In conclusion the IVS30+1G>T not only leads to a severe intracitoplasmatic alterations, a very enlarge endoplasmatic reticulum, but also to the alteration of mRNA expression of specific thyroid genes in the nodular and non-nodular goitrous tissues. The clinical evolution of patients harboring this mutations strengthen the concept of the influence of the environment, like iodine nutrition, to determine the final phenotypic appearance.
Acknowledgments: We acknowledge the financial support of Fapesp (grant 03/02989-8) and Instituto da Tiróide, São Paulo, Brazil. No other potencial conflict of interest relevant to this article was report.
Received in 2/9/2008
Accepted in 9/9/2008
- 1. Loeber JG. Neonatal screening in Europe: the situation in 2004. J Inherit Metab Dis. 2007;30(4):430-8.
- 2. Knobel M, Medeiros-Neto G. An outline of inherited disorders of the thyroid hormone generating system. Thyroid. 2003;13(8): 771-801.
- 3. Caputo M, Rivolta CM, Esperante SA, Gruñeiro-Papendieck L, Chiesa A, Pellizas CG, et al. Congenital hypothyroidism with goitre caused by new mutations in the thyroglobulin gene. Clin Endocrinol. 2007;67(3):351-7.
- 4. Ieiri T, Cochaux P, Targovnik HM, Suzuki M, Shimoda S, Perret J, et al. A 3′ splice site mutation in the thyroglobulin gene responsible for congenital goiter with hypothyroidism. J Clin Invest. 1991;88:1901-5.
- 5. Rivolta CM, Targovnik HM. Molecular advances in thyroglobulin disorders. Clin Chim Acta. 2006;374(1-2):8-24.
- 6. Targovnik H, Vono J, Billerbeck AEC, Cerrone GE, Varela V, Mendive F, et al. A 138-nucleotide deletion in the thyroglobulin ribonucleic acid messenger in a congenital goiterwith defective thyroglobulin synthesis. J Clin Endocrinol Metab. 1995;80: 3356-60.
- 7. Targovnik HM, Rivolta CM, Mendive FM, Moya CM, Medeiros-Neto G. Congenital goiter with hypothyroidism caused by a 5′ Splice Site mutation in the thyroglobulin gene. Thyroid. 2001; 11:685-90.
- 8. Pardo V, Rubio IG, Knobel M, Aguiar-Oliveira MH, Santos MM, Gomes SA, et al. Phenotypic variation among four family members with congenital hypothyroidism caused by two distinct thyroglobulin gene mutations. Thyroid. 2008;18(7):783-6.
- 9. Medeiros-Neto G, Bunduki V, Tomimori E, Gomes S, Knobel M, Martin RT, et al. Clinical case seminar: Prenatal Diagnosis and Treatment of Dyshormonogenetic Fetal Goiter Due to Defective Thyroglobulin Synthesis. J Clin Endocri Metab. 1997;82:4239-42.
- 10. Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26(6):509-15.
- 11. Castro MR, Bergert ER, Beito TG, McIver B, Goellner JR, Morris JC. Development of monoclonal antibodies against the human sodium iodide symporter: immunohistochemical characterization of this protein in thyroid cells. J Clin Endocrinol Metab. 1999;84:2957-62.
- 12. Medeiros-Neto G, Kim PS, Yoo SE, Vono J, Targovnik HM, Camargo R, et al. Congenital hypothyroid goiter with deficient thyroglobulin. Identification of an endoplasmic reticulum storage disease with induction of molecular chaperones. J Clin Invest. 1996;98:2838-44.
- 13. Camargo R, Limbert E, Gillam M, Henriques MM, Fernandes C, Catarino AL, et al. Aggressive metastatic follicular thyroid carcinoma with anaplastic transformation arising from a long-standing goiter in a patient with Pendred's syndrome. Thyroid. 2001;11(10):981-8.
- 14. Alzahrani AS, Baitei EY, Zou M, Shi Y. Clinical case seminar: metastatic follicular thyroid carcinoma arising from congenital goiter as a result of a novel splice donor site mutation in the thyroglobulin gene. J Clin Endocrinol Metab. 2006;91(3):740-6.
- 15. Hishinuma A, Fukata S, Kakudo K, Murata Y, Ieiri T. High incidence of thyroid cancer in long-standing goiters with thyroglobulin mutations. Thyroid. 2005;15(9):1079-84.
- 16. Vono-Toniolo J, Rivolta CM, Targovnik HM, Medeiros-Neto G, Kopp P. Naturally occurring mutations in the thyroglobulin gene. Thyroid. 2005;15(9):1021-33.
- 17. van de Graaf SA, Ris-Stalpers C, Pauws E, Mendive FM, Targovnik HM, de Vijlder JJ. Up to date with human thyroglobulin. J Endocrinol. 2001;170(2):307-21.
- 18. Yang SX, Pollock HG, Rawitch AB. Glycosylation in human thyroglobulin: location of the N-linked oligosaccharide units and comparison with bovine thyroglobulin. Arch Biochem Biophys. 1996;327(1):61-70.
- 19. Bruno R, Ferretti E, Tosi E, Arturi F, Giannasio P, Mattei T, et al. Modulation of thyroid-specific gene expression in normal and nodular human thyroid tissues from adults: an in vivo effect of thyrotropin. J Clin Endocrinol Metab. 2005;90(10):5692-7.
- 20. Lazar V, Bidart JM, Caillou B, Mahé C, Lacroix L, Filetti S, et al. Expression of the Na+/I- symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J Clin Endocrinol Metab. 1999;84(9):3228-34.
- 21. Lacroix L, Michiels S, Mian C, Arturi F, Caillou B, Filetti S, et al. HEX, PAX-8 and TTF-1 gene expression in human thyroid tissues: a comparative analysis with other genes involved in iodide metabolism. Clin Endocrinol (Oxf). 2006;64(4):398-404.
- 22. Mian C, Lacroix L, Alzieu L, Nocera M, Talbot M, Bidart JM, et al. Sodium iodide symporter and pendrin expression in human thyroid tissues.Thyroid. 2001;11(9):825-30.
- 23. Ohno M, Zannini M, Levy O, Carrasco N, di Lauro R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol. 1999;19(3):2051-60.
- 24. Dentice M, Luongo C, Elefante A, Ambrosio R, Salzano S, Zannini M, et al. Pendrin is a novel in vivo downstream target gene of the TTF-1/Nkx-2.1 homeodomain transcription factor in differentiated thyroid cells. Mol Cell Biol. 2005;25(22):10171-82.
- 25. Akagi T, Luong QT, Gui D, Said J, Selektar J, Yung A, et al. Induction of sodium iodide symporter gene and molecular characterisation of HNF3beta/FoxA2, TTF-1 and C/EBPbeta in thyroid carcinoma cells. Br J Cancer. 2008 [Epub ahead of print]
- 26. Kambe F, Seo H. Thyroid-specific transcription factors. Endocr J. 1997;44(6):775-84.
- 27. Damante G, Tell G, Di Lauro R. A unique combination of transcription factors controls differentiation of thyroid cells. Prog Nucleic Acid Res Mol Biol. 2001;66:307-56.
- 28. Zannini M, Francis-Lang H, Plachov D, Di Lauro R. Pax-8, a paired domain-containing protein, binds to a sequence overlapping the recognition site of a homeodomain and activates transcription from two thyroid-specific promoters. Mol Cell Biol. 1992;12(9):4230-41.
- 29. Damante G. Thyroid defects due to Pax8 gene mutations. Eur J Endocrinol. 1998;139(6):563-6.
- 30. Gérard AC, Daumerie C, Mestdagh C, Gohy S, De Burbure C, Costagliola S, et al. Correlation between the loss of thyroglobulin iodination and the expression of thyroid-specific proteins involved in iodine metabolism in thyroid carcinomas. J Clin Endocrinol Metab. 2003;88(10):4977-83.
- 31. Sodré AK, Rubio IG, Galrão AL, Knobel M, Tomimori EK, Alves VA, et al. Association of Low Sodium-Iodide Symposter mRNA Expression in Malignant Thyroid Nodules with Increased Intracellular Protein Staining. J Clin Endocrinol Metab. 2008 [Epub ahead of print]
- 32. Dohan O, Baloch Z, Banrevi Z, Livolsi V, Carrasco N. Predominant intracellular overexpression of the Na(+)/I(-) symporter (NIS) in a large sampling of thyroid cancer cases. J Clin Endocrinol Metab. 2001;86:2697-700.
- 33. Vono J, Lima N, Knobel M, Medeiros-Neto G. The effect of oral administration of iodine to patients with goiter and hypothyroidism due to defective synthesis of thyroglobulin. Thyroid. 1996;6(1):11-5.
- 34. Piosik PA, van Groenigen M, van Doorn J, Baas F, de Vijlder JJ. Effects of maternal thyroid status on thyroid hormones and growth in congenitally hypothyroid goat fetuses during the second half of gestation. Endocrinology. 1997;138(1):5-11.
Publication Dates
-
Publication in this collection
16 Jan 2009 -
Date of issue
Nov 2008
History
-
Accepted
09 Sept 2008 -
Received
02 Sept 2008