Abstracts
Many cases have been published showing a co-existence of autoimmune thyroid diseases (AITDs) and other autoimmune diseases. About a quarter of patients with achalasia have a concurrent thyroid disease, most commonly associated with hypothyroidism. Although relatively rare, the association of achalasia and hyperthyroidism requires attention. The physiopathology of Grave's Disease (GD) involves B- and T-mediator lymphocytes, which have an affinity for known thyroid antigens: thyroglobulin, thyroid-peroxidase, and thyrotrophin receptor. Currently, however, the real physiopathogenesis of achalasia continues to be unknown. Some important findings are suggestive of an autoimmune mechanism: significant infiltration of the myoenteric plexus by monocytes, presence of the class II-Human Histocompatibility Complex DQwl antigen and antibodies to myoenteric neurons. The present case reports a patient who, despite testing negative for Chagas' disease, had achalasia, progressed to developing significant wasting and worsening of his quality of life, was later diagnosed with hyperthyroidism. After endoscopic esophageal dilatation and radioiodine ablation of the thyroid gland, there was great improvement in the patient clinical condition. Arq Bras Endocrinol Metab. 2012;56(9):677-82
Muitos casos têm sido publicados mostrando uma coexistência entre as doenças autoimunes da tireoide (DAIT) e outras doenças autoimunes. Cerca de um quarto dos pacientes com acalasia têm doenças da tireoide concomitantemente, sendo a mais comum a associação com hipotireoidismo. Apesar de ser relativamente rara, a associação da acalasia e hipertireoidismo requer atenção. A fisiopatologia da doença de Graves (DG) envolve os linfócitos B e T-mediados, os quais têm afinidade pelos antígenos da tireoide: tireoglobulina, tireoperoxidase e receptor de tireotrofina. Atualmente, a real fisiopatogenia da acalasia continua desconhecida. No entanto, alguns importantes achados em análise são sugestivos de mecanismo autoimune: infiltração significativa do plexo mioentérico pelos monócitos, presença do antígeno-DQwl do Complexo Humano de Histocompatibilidade classe II e presença de anticorpos contra neurônios mioentéricos. Este presente caso aborda um paciente que, apesar de testes negativos para doença de Chagas, tem acalasia que progrediu para o desenvolvimento de significativa perda ponderal e piora da sua qualidade de vida, posteriormente, diagnosticado com hipertireoidismo. Após dilatação endoscópica esofágica e ablação da glândula tireoide com radioiodo, houve grande melhora na condição clínica do paciente. Arq Bras Endocrinol Metab. 2012;56(9):677-82
CASE REPORT
Achalasia and thyroid disease: possible autoimmune connection?
Acalasia e doença tireoidiana: uma conexão autoimune possível?
Ana Rosa P. QuiduteI,II; Eduardo Vasconcelos de FreitasI; Tadeu Gonçalves de LimaI; Ana Márcia Lima FeitosaI; Joyce Paiva dos SantosI; José Walter CorreiaI
IInternal Medicine Unit of the Teaching General Public Health Hospital Geral Dr. Cesar Calls (HGCC), Fortaleza, CE, Brazil
IIEndocrinology Unit of University Hospital Walter Cantídio, Universidade Federal do Ceará, Fortaleza, CE, Brazil
Correspondence Correspondence to: Ana Rosa P. Quidute Av. Padre Antonio Tomas, 3535 60190-020 Fortaleza, CE, Brazil arquidute@gmail.com
SUMMARY
Many cases have been published showing a co-existence of autoimmune thyroid diseases (AITDs) and other autoimmune diseases. About a quarter of patients with achalasia have a concurrent thyroid disease, most commonly associated with hypothyroidism. Although relatively rare, the association of achalasia and hyperthyroidism requires attention. The physiopathology of Grave's Disease (GD) involves B- and T-mediator lymphocytes, which have an affinity for known thyroid antigens: thyroglobulin, thyroid-peroxidase, and thyrotrophin receptor. Currently, however, the real physiopathogenesis of achalasia continues to be unknown. Some important findings are suggestive of an autoimmune mechanism: significant infiltration of the myoenteric plexus by monocytes, presence of the class II-Human Histocompatibility Complex DQwl antigen and antibodies to myoenteric neurons. The present case reports a patient who, despite testing negative for Chagas' disease, had achalasia, progressed to developing significant wasting and worsening of his quality of life, was later diagnosed with hyperthyroidism. After endoscopic esophageal dilatation and radioiodine ablation of the thyroid gland, there was great improvement in the patient clinical condition. Arq Bras Endocrinol Metab. 2012;56(9):677-82
SUMÁRIO
Muitos casos têm sido publicados mostrando uma coexistência entre as doenças autoimunes da tireoide (DAIT) e outras doenças autoimunes. Cerca de um quarto dos pacientes com acalasia têm doenças da tireoide concomitantemente, sendo a mais comum a associação com hipotireoidismo. Apesar de ser relativamente rara, a associação da acalasia e hipertireoidismo requer atenção. A fisiopatologia da doença de Graves (DG) envolve os linfócitos B e T-mediados, os quais têm afinidade pelos antígenos da tireoide: tireoglobulina, tireoperoxidase e receptor de tireotrofina. Atualmente, a real fisiopatogenia da acalasia continua desconhecida. No entanto, alguns importantes achados em análise são sugestivos de mecanismo autoimune: infiltração significativa do plexo mioentérico pelos monócitos, presença do antígeno-DQwl do Complexo Humano de Histocompatibilidade classe II e presença de anticorpos contra neurônios mioentéricos. Este presente caso aborda um paciente que, apesar de testes negativos para doença de Chagas, tem acalasia que progrediu para o desenvolvimento de significativa perda ponderal e piora da sua qualidade de vida, posteriormente, diagnosticado com hipertireoidismo. Após dilatação endoscópica esofágica e ablação da glândula tireoide com radioiodo, houve grande melhora na condição clínica do paciente. Arq Bras Endocrinol Metab. 2012;56(9):677-82
INTRODUCTION
Autoimmune thyroidopathies affect in average 2% to 5% of the general population, with young adult females and the elderly being the most susceptible patients. In these groups, Graves' disease (GD) hyperthyroidism and Hashimoto's thyroiditis causing hypothyroidism are the most prevalent etiologies. Although genetics is well known to cause and influence the progression of autoimmune diseases in approximately 79% (1), other environmental factors are known to be involved in the development of autoimmune thyroid diseases: quantity of ingested iodine, stress, drugs, pregnancy and changes in sexual hormones (1,2).
Graves' disease is the most common type of autoimmune hyperthyroidism, corresponding to approximately 60%-80% of total cases, affecting women aged 20-50 years. Since close to 50% of GD cases do not manifest concomitant Graves' ophthalmopathy, complete diagnosis is less obvious (2,3).
The physiopathology of GD involves the B and T-mediator lymphocytes, which have an affinity for known thyroid antigens: thyroglobulin, thyroid-peroxidase and thyrotrophin receptor. However, the thyrotrophin receptor is by itself a primary autoantigen in GD and is responsible for the hyperthyroidism manifestations of the disease. Varied genes with significant susceptibility to autoimmune diseases involving the thyroid gland have been identified and classified into two main clusters: gene immune-regulators (HLA, CTLA4, PTPN22, CD40, CD25, and FCRL3), and specific thyroid genes (thyroglobulin, TSH-receptor). It is well-known that they increase the susceptibility to other autoimmune diseases (1,4-6).
Many cases have been published showing a co-existence between autoimmune thyroid diseases (AITDs) and other autoimmune diseases for instance: systemic erythematous lupus, Sjögren syndrome, pernicious anemia, primary biliary cirrhosis, vitiligo, and type 1 diabetes mellitus (4). It is important to note that the immune-pathophysiological process involving AITDs, with emphasis on GD, is similar to that identified in other autoimmune diseases: cardinal participation of antibodies. Thus, as expected, target organs suffer significant lymphocyte infiltration in AITDs, with associated activated T and B-lymphocytes. It seems that GD pathogenesis results from both a central and peripheral dysfunctional tolerance homeostasis of the immune system. Thus a continuous initiated autoimmune process is propagated and maintained by a clone of T and B-cells (3).
Esophageal achalasia is a rare primary dysfunction involving the physiology motility of the esophageal body and malfunction of the lower esophageal sphincter (LES) characterized by respective progressive deficient peristalsis and incomplete relaxation when swallowing. Consequently, progressive obstruction develops at the esophageal-stomach junction, with associated proximal esophageal dilation (7,8).
Kraichely and cols. demonstrated that 25.7% of the patients with primary achalasia had one or more neural autoantibodies: skeletal muscle AChR or striational neuronal voltagegated cation channel (potassium channel or N-type calcium channel) or glutamic acid decarboxylase-65 antibody (GAD65). By contrast, only 4.4% of healthy subjects had one or more of these autoantibodies. This profile of autoantibodies suggests an autoimmune basis for a subset of primary achalasia (9).
The pathophysiology of achalasia involves the progressive degeneration of intrinsic neurons found in the esophageal wall, with a significant decrease in the population of functional ganglia of the myoenteric plexus. Besides, the remaining ganglia are usually invaded by countless lymphocytes and to lesser degree, eosinophils. Moreover, this inflammatory degradation involves mainly nitric oxide-producing inhibitory neurons, consequently affecting the relaxation capacity of the smooth muscle of LES; while the involvement of cholinergic neurons results in an abnormally high resting tonus, making it progressively difficult for physiologic contraction to occur (8). It is well known that the resting tonus of the LES is regulated by excitatory (acetylcholine, Substance P, among others), as well as inhibitory neurotransmitters (nitric oxide). Therefore, patients with achalasia have a significant imbalance between the excitatory and inhibitory pathways, resulting in hypertensive LES (10). Currently, the real physiopathogenesis of achalasia continues to be unknown. However, some important findings are suggestive of an autoimmune mechanism: significant infiltration of the myoenteric plexus by monocytes, presence of the class II-Human Histocompatibility Complex DQwl antigen and antibodies to myoenteric neurons. Specifically, higher genotypic frequency of the human leukocyte antigen (HLA)-DQw1, DQA1*0101, DQA1*103, DQB1*0602, and DQB1*0603 alleles were observed in achalasia patients compared with controls (7,8,10-13). Descriptions of inflammatory infiltration in the affected regions of the esophagus in achalasia led to speculation of an autoimmune pathogenesis. This was reinforced by a histochemical study that showed predominant T- and B-cell lymphocytic infiltrates along the nerve fascicles and around ganglion cells, supporting the concept of an inflammatory, probably autoimmune, etiology of autonomic nervous system injury in patients with achalasia (14,15).
Storch and cols. demonstrated IgG-antibodies against Auerbach's plexus by standard indirect immunofluorescence. Antibodies to the cytoplasm of Auerbach's plexus were found in 37 of 58 patients with achalasia at variable stages of the disease (I-IV) with a disease duration ranging from 1 to 20 years (16). Despite the fact that these findings link a possible autoimmune mechanism to achalasia, further studies are needed to establish if autoimmunity is a primary etiology or a co-factor in the pathogenesis of achalasia.
Frequently, patients with goiter refer to dysphagia which, in many cases, is attributed to mechanical factors caused by the enlarged gland. Unfortunately, clinical investigations usually ignore the existence of achalasia as the cause of dysphagia, despite the fact there is sufficient literature citations linking it mainly to Hashimoto thyroiditis and, to a lesser degree, to hyperthyroidism (17).
This case report the disease history of a patient who had achalasia, and was later diagnosed with hyperthyroidism due to GD. Our main objective is to discuss such a rare association, citing the possible participation of autoimmunity and the conduct followed to improve the quality of life of the patient.
CASE REPORT
A.M.A., a 57-year-old male, coming from a metropolis non-endemic for Chagas' disease, was admitted to our hospital reporting frequent vomiting and significant weight loss. Over the last 7 months, he had a sensation of 'dry mouth' and 'burning' in the anterior cervical region, which extended up to the epigastric region. It was characteristically worse in the morning after waking up, but improved with the ingestion of refrigerated liquids. Three months before admission, daily post-prandial vomiting (4 to 5 times) became frequent, besides considerable weight loss (35%; from 97 kg to 62.5 kg in three months), and progressive salivation, dysphagia, and general lethargy. No fever was reported during this period. During physical examination, low body weight was remarkable, together with painful abdomen upon superficial and profound palpation around the epigastric and mesogastric region. The thyroid gland was painless and appeared diffusely enlarged, with its volume approximately three times greater than normal and fibrous-elastic texture. This was later confirmed by a cervical ultrasound that showed diffuse goiter. Other findings of the physical examination were unremarkable.
An upper digestive endoscopy and esophagogram were carried out, owing to the predominant dysphagy-related symptoms, both suggestive of esophageal achalasia involving the lower esophageal sphincter (LES). Due to the notable diffuse goiter upon physical examination, we dosed thyroid-related hormones, and the results were compatible with primary hyperthyroidism (Table 1).
Laboratory findings and physical analysis were determinant for the indication of a thyroid scintigraphy followed by 131I ablation therapy. In relation to significant body weight loss and signs of malnutrition, we carried out endoscopic esophageal dilation, so as to improve his nutritional status. His epigastric pain owing to achalasia was treated conservatively, postponing Heller's cardiomiotomy. As a result, the patient evolved with complete remission of his symptoms, including the vomiting episodes. He was discharged from the hospital, and electively referred to the general surgery department, where he referred to heartburn and early abdominal satiety, but no vomiting since dilation was carried out. Furthermore, during this extra-hospital period, he recovered his body weight remarkably (62.5 kg to a current 92.6 kg, in one year), which led us to conclude that clinical treatment improved significantly his nutritional status and quality of life. This was the reason for us to avoid Heller's cardiomiotomy.
DISCUSSION
The cause for the inflammatory degeneration of esophageal neurons seen in achalasia is yet to be elucidated. The most accepted physiopathological hypothesis in idiopathic achalasia is a decrease in myenteric plexus neurons in the esophagus. This alteration in the myenteric plexus has been explained by different etiopathogenic mechanisms: infection, genetics, autoimmunity, ischemia, toxicicity etc. In recent years, the hypothesis of an autoimmune cause has acquired special relevance based on three aspects: histopathological findings (presence of mononuclear and lymphocyte infiltration in the myenteric plexus in esophageal tissue samples of patients with achalasia), the association between class II antigen of the major complex of histocompatibility HLA-DQw1 and non-familial idiopathic achalasia; and the presence of autoantibodies against the myenteric plexus (AAM) in the serum of a subgroup of patients with achalasia (8-10,13). A cluster consisting of varied autoimmune diseases has been linked to achalasia: myasthenia gravis, polymyositis, autoimmune thyroid disease, among others, contributing to the argument that autoimmunity may be a cause for some idiopathic achalasia (17-19). A further achalasia candidate gene selected based on its involvement in autoimmunity is the protein of tyrosine phosphatase N22 gene (PTPN22) on chromosome 1p13 (20). PTPN22 encodes a lymphoid-specific phosphatase (LYP) that downregulates T-cell activation. It has been postulated that the PTPN22-allele 1858T promotes an autoimmune response that results in chronic inflammation and the associations with some autoimmune conditions have already been established. A gender-specific association has been observed between the T allele of C1858T and achalasia in female patients (20,21).
Recently, Nunez and cols. described that variants in PTPN22 and IL23R genes confer predisposition to develop achalasia (22). Because these polymorphisms are common risk factors to several immunologic disorders, their results confirm that genetic variants that predispose to these diseases may also confer susceptibility to achalasia. This finding supports the hypothesis that achalasia shares molecular pathways with other immune-mediated diseases, which could explain the complexity of pathogenesis and progression of the disease (22). However, we could not conduct a genetic study, which we believe can provide a sample set of patient at high-risk of developing achalasia from the general population in the near future.
In our setting, the greatest prevalence of megaesophagus is due to Chagas' disease, thus this disease is the main differential diagnosis. Diagnosis in the chronic phase of Chagas' disease is essentially serological; indirect hemagglutination tests, ELISA, and indirect immunofluorescence can determine the diagnosis in virtually 100% of cases (23). However, our patient tested negative for Chagas' disease and this etiology was ruled out. Furthermore, he denied any contact with the prevalent vector of the disease, came from a metropolis not endemic for Chagas' and did not have previous history of blood transfusions (23).
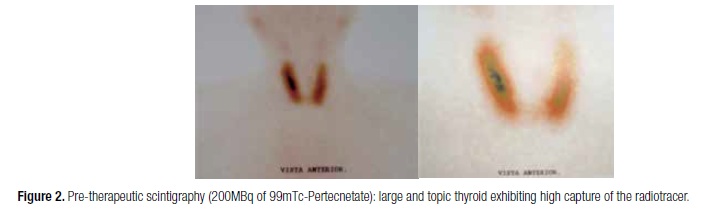
We are aware of varied causes of hyperthyroidism. The diagnosis of GD was determined by: suppressed TSH, increased free T4 and T3, a diffuse goiter determined by ultrasonography and the capture of the radiotracer by the thyroid gland in a diffuse pattern. These findings reduced the chances of attributing thyroid disease to sub-acute thyroiditis or toxic uni- or multi-nodular goiter, reinforcing the autoimmune etiology. Furthermore, the fact that physical examination showed no Graves' ophthalmopathy did not rule out the disease, since up to 50% of diagnosed patients do not manifest ocular disease in their presentation of hyperthyroidism (3). Intolerance to glucose seen in our patient may be attributed to uncontrolled hyperthyroidism, since fasting levels fell to normal after treatment and outpatient follow-up (24).
The association between achalasia and AITDs has been described in literature. However, little is taken into consideration when dealing with a patient with goiter and who refers to achalasia-like symptoms, usually due to the extrinsic compression of the hypertrophied gland on the esophagus.
Emami and cols. (17), examining a group of 30 patients (20 women and 10 men) with achalasia, whose ages varied from 30 to 70 years, found that this esophageal anomaly was associated with AITDs in 23% of the cases (17). In this group, hypothyroidism due to Hashimoto thyroiditis was the most common occurrence.
GD has been associated with achalasia in an infant who had to be subjected to Heller's cardiotomy (25). In young patients, it is worth having Allgrove or Triple A syndrome (primary adrenal insufficiency, achalasia, and alagrimia) as a differential diagnosis (10). Despite the age, primary adrenal insufficiency was investigated in our patient and ruled out.
Usually, hyperthyroidism due to GD is diagnosed after a thorough symptomatic history and subsequent physical examination. However, our patient had no significant history, neither were the signs vivid, apart from a 'goiter-form' neck, which prompted us to investigate thyroid function. However, in their series, Emami and cols. (17) did not find a direct correlation between the gravity of thyroid disease and the degree of achalasia symptoms. This finding was similar in our case, where the patient had more significant esophageal symptoms than those related to the thyroid disease. In our case, we opted for primary treatment with I131, since plasma hormone concentrations were moderately high. Besides, the coexistence of untreated achalasia would be an impediment to treat him with conventional anti-thyroid drugs, due to increased difficulty in absorption. This serves as an alert to clinicians and specialists when treating patients with GD with resistance to conventional treatment.
In conclusion, achalasia may be associated with varied autoimmune thyroid diseases (hyperthyroidism, hypothyroidism and thyroid nodular diseases) that ought to be considered when a new case of idiopathic achalasia is diagnosed. Additionally, patients presenting any autoimmune thyroid disease should be further investigated for esophagus-related problems, ranging from characteristic esophageal dysmotility to more complex situations, such as excessive weight loss, that cannot be explained by thyroid disease, as occurred in our case.
Moreover, antibodies to the myoenteric complex suggest autoimmune mechanisms. Probably, an unknown environmental insult may determine inflammation of the plexus in a genetically susceptive person, with progressive association with thyroid gland involvement. Therefore, autoimmunity may either be a co-factor or aid the pathogenesis of signs and symptoms (10). However, controversies continue to exist: whether we are dealing with a true cause-effect situation or a simple epiphenomenon. This sparks the need for further investigative immunopathological studies, explaining the cause-effect relationship in these diseases.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received on July/28/2012
Accepted on Oct/2/2012
- 1. Davies TF, Latif R, Yin X. New genetic insights from autoimmune thyroid disease. J Thyroid Res. 2012;2012:623852.
- 2. Weetman AP. Graves' disease. N Engl J Med. 2000;343(17):1236-48.
- 3. Seigel SC, Hodak SP. Thyrotoxicosis. Med Clin North Am. 2012;96(2):175-201.
- 4. Weetman AP. Diseases associated with thyroid autoimmunity: explanations for the expanding spectrum. Clin Endocrinol (Oxf). 2011;74(4):411-8.
- 5. Sibarani RP. Genetics of Graves' disease: the lost concept. Acta Med Indones. 2009;41(1):37-40.
- 6. Yin X, Latif R, Bahn RS, Davies TF. Genetic Profiling in Graves' Disease: Further Evidence for Lack of a Distinct Genetic Contribution to Graves' Ophthalmopathy. Thyroid. 2012.
- 7. Eckardt AJ, Eckardt VF. Current clinical approach to achalasia. World J Gastroenterol. 2009;15(32):3969-75.
- 8. Francis DL, Katzka DA. Achalasia: update on the disease and its treatment. Gastroenterology. 2010;139(2):369-74.
- 9. Kraichely RE, Farrugia G, Pittock SJ, Castell DO, Lennon VA. Neural autoantibody profile of primary achalasia. Dig Dis Sci. 2010;55(2):307-11.
- 10. Kraichely RE, Farrugia G. Achalasia: physiology and etiopathogenesis. Dis Esophagus. 2006;19(4):213-23.
- 11. Wong RK, Maydonovitch CL, Metz SJ, Baker JR Jr. Significant DQw1 association in achalasia. Dig Dis Sci. 1989;34(3):349-52.
- 12. Verne GN, Hahn AB, Pineau BC, Hoffman BJ, Wojciechowski BW, Wu WC. Association of HLA-DR and -DQ alleles with idiopathic achalasia. Gastroenterology. 1999;117(1):26-31.
- 13. Ghoshal UC, Daschakraborty SB, Singh R. Pathogenesis of achalasia cardia. World J Gastroenterol. 2012;18(24):3050-7.
- 14. Gockel I, Bohl JR, Doostkam S, Eckardt VF, Junginger T. Spectrum of histopathologic findings in patients with achalasia reflects different etiologies. J Gastroenterol Hepatol. 2006;21(4):727-33.
- 15. Raymond L, Lach B, Shamji FM. Inflammatory aetiology of primary oesophageal achalasia: an immunohistochemical and ultrastructural study of Auerbach's plexus. Histopathology. 1999;35(5):445-53.
- 16. Storch WB, Eckardt VF, Wienbeck M, Eberl T, Auer PG, Hecker A, et al. Autoantibodies to Auerbach's plexus in achalasia. Cell Mol Biol (Noisy-le-grand). 1995;41(8):1033-8.
- 17. Emami MH, Raisi M, Amini J, Daghaghzadeh H. Achalasia and thyroid disease. World J Gastroenterol. 2007;13(4):594-9.
- 18. Domsic R, Fasanella K, Bielefeldt K. Gastrointestinal manifestations of systemic sclerosis. Dig Dis Sci. 2008;53(5):1163-74.
- 19. Booy JD, Takata J, Tomlinson G, Urbach DR. The prevalence of autoimmune disease in patients with esophageal achalasia. Dis Esophagus. 2012;25(3):209-13.
- 20. Gockel HR, Schumacher J, Gockel I, Lang H, Haaf T, Nothen MM. Achalasia: will genetic studies provide insights? Hum Genet. 2010;128(4):353-64.
- 21. Santiago JL, Martinez A, Benito MS, Ruiz de Leon A, Mendoza JL, Fernandez-Arquero M, et al. Gender-specific association of the PTPN22 C1858T polymorphism with achalasia. Hum Immunol. 2007;68(10):867-70.
- 22. Nunez C, Garcia-Gonzalez MA, Santiago JL, Benito MS, Mearin F, de la Concha EG, et al. Association of IL10 promoter polymorphisms with idiopathic achalasia. Hum Immunol. 2011;72(9):749-52.
- 23. Saúde SdVe, Saúde Md. Consenso Brasileiro em Doença de Chagas. Rev Soc Bras Med Trop. 2005;38.
- 24. Kabadi UM, Eisenstein AB. Glucose intolerance in hyperthyroidism: role of glucagon. J Clin Endocrinol Metab. 1980;50(2):392-6.
- 25. Srinivasan R, Cosgrove M, Huddart S, Evans D. Autoimmune thyrotoxicosis with achalasia cardia. Indian J Pediatr. 2009;76(8):850-1.
Correspondence to:
Publication Dates
-
Publication in this collection
15 Jan 2013 -
Date of issue
Dec 2012
History
-
Received
28 July 2012 -
Accepted
02 Oct 2012