Abstracts
Patients with adult onset non-familial progressive ataxia are classified in sporadic ataxia group. There are several disease categories that may manifest with sporadic ataxia: toxic causes, immune-mediated ataxias, vitamin deficiency, infectious diseases, degenerative disorders and even genetic conditions. Considering heterogeneity in the clinical spectrum of sporadic ataxias, the correct diagnosis remains a clinical challenge. In this review, the different disease categories that lead to sporadic ataxia with adult onset are discussed with special emphasis on their clinical and neuroimaging features, and diagnostic criteria.
ataxia; sporadic ataxia; clinical features; diagnostic criteria
Pacientes com ataxia progressiva que se inicia na idade adulta, e sem histórico familiar da doença, são classificados no grupo das ataxias esporádicas. Existem várias categorias de doenças que podem se manifestar com ataxia esporádica, tais como: causas tóxicas, ataxias imunomediadas, deficiência de vitaminas, doenças infecciosas, doenças degenerativas e até mesmo condições genéticas. Considerando a heterogeneidade no espectro clínico das ataxias esporádicas, a definição da etiologia constitui um desafio diagnóstico. Neste artigo de revisão, realizamos uma discussão sobre as diferentes categorias de doenças que causam ataxia com início na idade adulta sem histórico familiar, com ênfase nas características clínicas, aspectos de imagem e critérios diagnósticos.
ataxia; ataxias esporádicas; aspectos clínicos; critérios diagnósticos
The term ataxia traditionally means loss of order, and clinically denotes loss of balance and coordination. On the other hand, the name sporadic means without spot or regular pattern. Based on that, sporadic ataxia is defined as a neurological condition characterized by ataxia of adult onset, usually over 40 years old, with family history negative for ataxia or its related symptoms and signs 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. .
The terminology ‘cerebellar ataxias’ comprises a wide spectrum of neurological disorders with ataxia as the main symptom. Ataxia results from the involvement of cerebellar structures, or from a combination of cerebellar and extra-cerebellar lesions, especially the brainstem
22 . Manto M, Marmolino D. Cerebellar ataxias. Current opinion in neurology. 2009;22:419-429.
. Also, ataxias are a heterogeneous group of diseases comprising genetic and non-genetic etiologies. According to current etiology-based classifications, the ataxias can be subdivided into six major groups: autosomal dominant spinocerebellar ataxias (SCA), autosomal recessive ataxias, congenital ataxias, mitochondrial ataxias, X-linked cerebellar ataxias and sporadic ataxias
11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104.
2 . Manto M, Marmolino D. Cerebellar ataxias. Current opinion in neurology. 2009;22:419-429.
-
33 . Mariotti C, Fancellu R, Di Donato S. An overview of the patient with ataxia. J Neurol 2005;252:511-518.
. A familial disorder affecting successive generations is suggestive of SCA, while in patients with an early disease onset, particularly before 25-year-old, an autosomal recessive inheritance is most likely
22 . Manto M, Marmolino D. Cerebellar ataxias. Current opinion in neurology. 2009;22:419-429.
,
33 . Mariotti C, Fancellu R, Di Donato S. An overview of the patient with ataxia. J Neurol 2005;252:511-518.
. However, patients with adult onset non-familial progressive ataxia are classified in sporadic ataxia group. There are several disease categories that may manifest with adult onset sporadic ataxia and owing to their heterogeneity, the correct diagnosis remains a clinical challenge
44 . Van Gaalen J, van de Warrenburg BP. A practical approach to late-onset cerebellar ataxia: putting the disorder with lack of order into order. Pract Neurol 2012;12:14-24.
.
In this review, the different disease categories that lead to sporadic ataxia with adult onset are discussed with special emphasis on their clinical features and diagnostic criteria. Purposely, stroke and demyelinating diseases causing ataxia were excluded from this review.
Alcoholic cerebellar degeneration
Alcoholic cerebellar degeneration is the most common form of acquired toxic ataxia and is a frequent neurological complication in alcoholics. Cerebellar symptoms usually begin in middle-aged, and there is a marked history of chronic alcohol abuse. Ataxia usually evolves rapidly, within weeks to months, but some patients have a slower progression 55 . Yokota O, Tsuchiya K, Terada S, et al. Frequency and clinicopathological characteristics of alcoholic cerebellar degeneration in Japan: a cross-sectional study of 1,509 postmortems. Acta Neuropathol 2006;112:43-51. . Neurological features comprise severe ataxia of gait and lower limbs with relatively mild involvement of the upper limbs. Interestingly, speech, ocular motility and coordination extremities are usually preserved. Also, signs of neuropathy, such as reduced or absent deep tendon reflexes, are frequent 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. , 55 . Yokota O, Tsuchiya K, Terada S, et al. Frequency and clinicopathological characteristics of alcoholic cerebellar degeneration in Japan: a cross-sectional study of 1,509 postmortems. Acta Neuropathol 2006;112:43-51. .
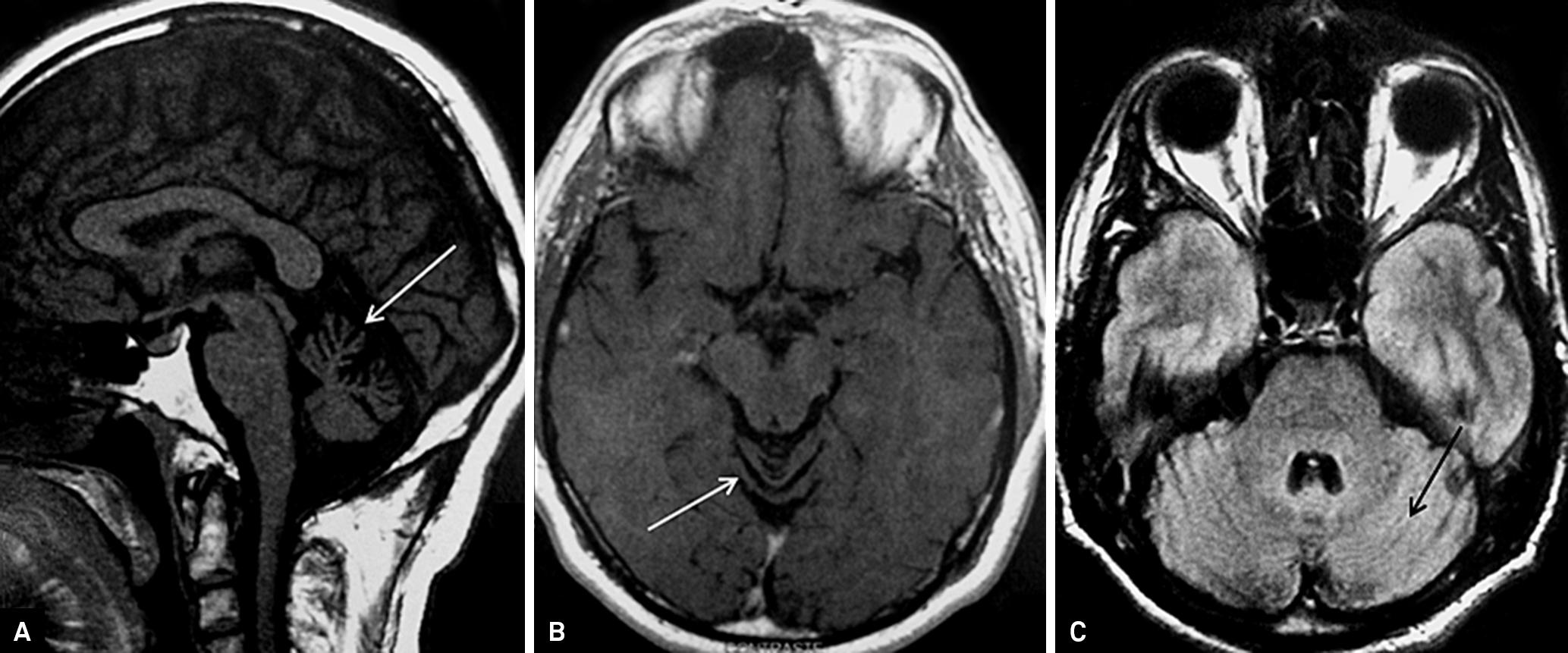
Neuroimaging features in alcoholic cerebellar degeneration typically demonstrate cerebellar atrophy, in general with preferential involvement of the superior cerebellar vermis ( Figure 1 ). Data from a clinicopathological study showed that cerebellar atrophy and degenerative process in alcoholic cerebellar degeneration begins in the superior vermis, and cerebellar hemispheres are less involved 55 . Yokota O, Tsuchiya K, Terada S, et al. Frequency and clinicopathological characteristics of alcoholic cerebellar degeneration in Japan: a cross-sectional study of 1,509 postmortems. Acta Neuropathol 2006;112:43-51. , 66 . Hillbom M, Muuronen A, Holm L, Hindmarsh T. The clinical versus radiological diagnosis of alcoholic cerebellar degeneration. J Neurol Sci 1986;73:45-53. . Curiously, another study showed that approximately 50% of the chronic alcohol users have cerebellar atrophy without ataxia. Symptomatic patients frequently have neuropsychological impairment and variable degrees of brain atrophy 55 . Yokota O, Tsuchiya K, Terada S, et al. Frequency and clinicopathological characteristics of alcoholic cerebellar degeneration in Japan: a cross-sectional study of 1,509 postmortems. Acta Neuropathol 2006;112:43-51. , 66 . Hillbom M, Muuronen A, Holm L, Hindmarsh T. The clinical versus radiological diagnosis of alcoholic cerebellar degeneration. J Neurol Sci 1986;73:45-53. .
. Brain MRI of a patient with alcoholic cerebellar degeneration (ACD). Sagittal T1-weighted brain MRI disclosed marked cerebellar atrophy restricted to the superior vermis (A). Axial T1-weighted MRI of the same patient confirming atrophy in the suoerior vermis (B). Axial FLAIR-weighted MRI: note that no atrophy in the medium or inferior vermis is observed (C). Neuroimaging features in ACD typically demonstrate cerebellar atrophy restricted to the superior cerebellar vermis.
The pathogenesis of alcoholic cerebellar degeneration includes both: the direct toxic effect of the alcohol to the cerebellum and the consequences of vitamin B1 (thiamine) deficiency. Alcohol produces a toxic effect to Purkinje cells, and also is responsible for inhibitory mechanisms in GABA system, increases lipid peroxidation, and reduces of antioxidant concentrations
77 . Torvik A, Torp S. The prevalence of alcoholic cerebellar atrophy. A morphometric and histological study of an autopsy material. J. Neurol Sci 1986;75:43-51.
8 . Jaatinen P, Rintala J. Mechanisms of ethanol-induced degeneration in the developing, mature, and aging cerebellum. Cerebellum 2008;7:332-347.
-
99 . Butterworth RF. Pathophysiology of alcoholic brain damage: synergistic effects of ethanol, thiamine deficiency and alcoholic liver disease. Metab Brain Dis 1995;10:1-8.
. Alcoholics have thiamine deficiency as a result of poor diet, gastrointestinal disorders and liver disease
11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104.
. Treatment of alcoholic cerebellar degeneration includes alcohol abstinence and thiamine replacement. Variable degrees of clinical response may occur with treatment: some patients have significant improvement of the ataxia; others have only stabilization of symptoms; while a smaller group may present slow progressive gait ataxia despite alcohol discontinuation
11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104.
.
Ataxia related to toxic causes
Besides alcohol, other substances abuse or exposure may also cause or exacerbate the already pre-existing ataxia. The main substances causing toxic ataxia are: lithium, phenytoin, amiodarone, toluene and some chemotherapy’s (5-fluorouracil and cytosine arabinoside)
1010 . Niethammer M, Ford B. Permanent lithium-induced cerebellar toxicity: three cases and review of literature. Mov Disord 2007;22:570-573.
11 . De Marcos FA, Ghizoni E, Kobayashi E, Li LM, Cendes F. Cerebellar volume and long-term use of phenytoin. Seizure 2003;12:312-315.
12 . Orr CF, Ahlskog JE. Frequency, characteristics, and risk factors for amiodarone neurotoxicity. Arch Neurol 2009;66:865-869.
13 . Fornazzari L, Wilkinson DA, Kapur BM, Carlen PL. Cerebellar, cortical and functional impairment in toluene abusers. Acta Neurol Scand 1983;67:319-329.
-
1414 . Bygrave HA, Geh JI, Jani Y, Glynne-Jones R. Neurological complications of 5-fluorouracil chemotherapy: case report and review of the literature. Clin Oncol (R Coll Radiol) 1998;10:334-336.
. There are also descriptions of ataxia induced by mercury or bismuth subsalicylate exposure
1515 . Larsen JO, Braendgaard H. Structural preservation of cerebellar granule cells following neurointoxication with methyl mercury: a stereological study of the rat cerebellum. Acta Neuropathol 1995;90:251-256.
,
1616 . Gordon MF, Abrams RI, Rubin DB, Barr WB, Correa DD. Bismuth toxicity. Neurology 1994;44:2418.
. Other drugs may rarely cause cerebellar symptoms: carbamazepin, valproic acid, cyclosporine, isoniazid, metronidazole, nitrofurantoin, procainamide and statins
1717 . Teive HA, Munhoz RP, Werneck LC. Acquired cerebellar ataxia due to statin use. Arq Neuropsiquiatr 2012;70:152.
,
1818 . Jain KK. Drug-induced cerebellar disorders. In: Jain KK (Ed.). Drug-induced neurological disorders, 1sted. Hogrefe and Huber Publishers, Seatle, 1996:275-282.
. When toxic ataxia is suspected, these substances should be immediately discontinued. Clinical outcome is variable after drug cessation: patients may have significant improvement or only stabilization of symptoms with permanent cerebellar atrophy
1818 . Jain KK. Drug-induced cerebellar disorders. In: Jain KK (Ed.). Drug-induced neurological disorders, 1sted. Hogrefe and Huber Publishers, Seatle, 1996:275-282.
.
Paraneoplastic cerebellar degeneration
Paraneoplastic cerebellar degeneration is an immune-mediated disorder that affects cerebellar cortex, usually associated with the following neoplastic diseases: small cell lung cancer, breast cancer, ovarian cancer and Hodgkin's lymphoma 1919 . Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008;7:327-340. , 2020 . Scaravilli F, An SF, Groves M, Thom M. The neuropathology of paraneoplastic syndromes. Brain Pathol 1999;9:251-260. . In paraneoplastic cerebellar degeneration the onset of gait ataxia is usually subacute, and symptoms usually develop in few weeks, with rapid progression 2121 . Shams’ili S, Grefkens J, de Leeuw B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain 2003;126:1409-1418. .
Some authors subdivide paraneoplastic cerebellar degeneration into four groups: 1- cerebellar ataxia associated with signs of a widespread involvement of the nervous system (encephalomyelitis): seizures, behavior changes, memory dysfunction, chorea, limbic encephalitis and peripheral neuropathy. This phenotype is usually related to high serum levels of anti-Hu and lung cancer; 2- pure cerebellar ataxia in women, associated with high serum levels of anti-Yo and gynecological tumors (breast or ovarian cancer); 3- pure cerebellar syndrome, more commonly observed in men, associated with Hodgkin's lymphoma and high serum levels of anti-Tr and mGluR1; 4- cerebellar ataxia associated with Lambert-Eaton syndrome related to high serum levels of anti-VGCC and lung cancer 1818 . Jain KK. Drug-induced cerebellar disorders. In: Jain KK (Ed.). Drug-induced neurological disorders, 1sted. Hogrefe and Huber Publishers, Seatle, 1996:275-282. , 2121 . Shams’ili S, Grefkens J, de Leeuw B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain 2003;126:1409-1418. , 2323 . Scheid R, Voltz R, Briest S, Kluge R, von Cramon DY. Clinical insights into paraneoplastic cerebellar degeneration. J Neurol Neurosurg Psychiatry 2006;77:529-530. .
Brain imaging may be frustrating, since initial magnetic resonance imaging (MRI) is usually unremarkable. Signal changes, particularly in superior vermis, may be found in the beginning of symptoms 2424 . Aragão M de M, Pedroso JL, Albuquerque MV, Dutra LA, Barsottini OG. Superior cerebellar hyperintense sign on FLAIR-weighted magnetic resonance imaging in paraneoplastic cerebellar degeneration. Arq Neuropsiquiatr 2012;70:967. . Cerebellar atrophy frequently develops in late stages. Cerebrospinal fluid (CSF) usually shows mild pleocytosis or high levels of proteins 2525 . De Andrés C, Esquivel A, de Villoria JG, Graus F, Sánchez-Ramón S. Unusual magnetic resonance imaging and cerebraspinal fluid findings in paraneoplastic cerebellar degeneration: a sequential study. J Neurol Neurosurg Psychiatry 2006;77:562-563. . Patients should be promptly screened for anti-neuronal antibodies, as described above. Search for the primary tumor is extremely relevant in this syndrome.
In general, patients with paraneoplastic cerebellar degeneration do not respond to treatment of the underlying tumor. Further, exceptional reports have demonstrated improvement of the ataxia symptoms after immunotherapy (intravenous human immunoglobulin, plasma exchange or steroids) 2626 . David YB, Warner E, Levitan M, Sutton DM, Malkin MG, Dalmau JO. Autoimmune paraneoplastic cerebellar degeneration in ovarian carcinoma patients treated with plasmapheresis and immunoglobulin. A case report. Cancer 1996;78:2153-2156. .
Immune-mediated ataxias
The cerebellum, in special the Purkinje cells, has an important immunological target in the context of some systemic diseases. Therefore, immune-mediated ataxias are neurological conditions related to auto-immune disorders. Immune-mediated ataxias include: gluten ataxia, ataxia associated with antibodies against the glutamic acid decarboxylase (anti-GAD), steroid-responsive encephalopathy associated with autoimmune thyroiditis and paraneoplastic cerebellar degeneration (described in the item above) 2727 . Hadjivassiliou M. Immune-mediated acquired ataxias. Handb Clin Neurol 2012;103:189-199. .
Anti-GAD ataxia
High levels of glutamic acid decarboxylase (GAD) autoantibody may be observed in a few patients with sporadic ataxias, supporting autoimmune pathogenesis of the cerebellar syndrome. The clinical spectrum of anti-GAD ataxia comprises slowly progressive cerebellar ataxia evolving in months or years, associated with cerebellar atrophy on brain MRI in about half of cases. Cerebrospinal fluid analysis may detect oligoclonal bands. Diagnosis is supported by high serum levels of GAD antibodies. Association with late-onset type 1 diabetes and other auto-immune disorders is may be observed 2828 . Fernandes M, Munhoz RP, Carrilho PE, et al. Neurological disorders associated with glutamic acid decarboxylase antibodies: a Brazilian series. Arq Neuropsiquiatr 2012;70:657-661. . Immunotherapy might be useful in anti-GAD ataxia, but this is still controversial 2929 . Pedroso JL, Braga-Neto P, Dutra LA, Barsottini OG. Cerebellar ataxia associated to anti-glutamic acid decarboxylase autoantibody (anti-GAD): partial improvement with intravenous immunoglobulin therapy. Arq Neuropsiquiatr 2011;69:993. .
Gluten related ataxia
Celiac disease is a chronic immune-mediated intestinal disease, characterized by inflammatory process and villous atrophy in intestinal mucosa. The diagnosis of celiac disease is based on typical lesions in intestinal biopsy, associated or not with specific antibodies. Much controversy remains about a real causal relationship between asymptomatic celiac disease and sporadic ataxia. Several data have demonstrated a strong association between cerebellar ataxia and silent coeliac disease, defined by the presence of antigliadin antibodies and tissue transglutaminase levels 3030 . Hadjivassiliou M, Grunewald RA, Chattopadhyay AK, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet 1998;352:1582-1585. . Researchers suggest that ataxia in patients with high serum levels of antigliadin and transglutaminase antibodies are immune-mediated and proposed the term “gluten ataxia". Antibodies against transglutaminase 6 (TG6) are gluten-dependent and appear to be a sensitive and specific marker of gluten ataxia. Of note, up to 12% of the healthy population may have high serum levels of antigliadin antibodies 3030 . Hadjivassiliou M, Grunewald RA, Chattopadhyay AK, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet 1998;352:1582-1585. , 3131 . Hadjivassiliou M, Sanders DS, Woodroofe N, Williamson C, Grünewald RA. Gluten ataxia. Cerebellum 2008;7:494-498. .
Steroid-responsive encephalopathy associated with autoimmune thyroiditis
Steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT), also known Hashimoto encephalopathy, is an immune-mediated disease characterized by subacute cognitive changes, cerebral ischemia, myoclonus, seizures, psychiatric symptoms, ataxia and high serum levels of antithyroid antibodies (thyperoxidase or thyroglobulin antibodies). Brain MRI may disclose mesial temporal lobe lesions, multiple subcortical ischemic areas or may be normal. CSF may show mild pleocytosis or high levels of proteins. Interestingly, neurological deficits have marked improvement after intravenous steroid therapy. Response to steroids and the presence of antithyroid antibodies, which are common markers of autoimmunity, suggest that SREAT is an immune-mediated disorder 3232 . Castillo P, Woodruff B, Caselli R, et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol 2006;63:197-202. .
Acquired ataxias due to vitamin deficiency
Vitamin B1 (Thiamine) deficiency
Vitamin B1 (thiamine) deficiency may cause ataxia in the context of Wernicke encephalopathy (ataxia, confusion and ophthalmoparesis), a condition more frequently observed in alcoholics. Also, Vitamin B1 deficiency is presumed to be involved in the pathophysiology of alcoholic cerebellar degeneration. Brain MRI may disclose symmetric signal changes in thalami, mamillary bodies, tectal plate and periaqueductal area 3333 . Spinazzi M, Angelini C, Patrini C. Subacute sensory ataxia and optic neuropathy with thiamine deficiency. Nat Rev Neurol 2010;6:288-293. .
Vitamin B12 deficiency
Vitamin B12 deficiency occurs in the setting of atrophic gastritis, pernicious anemia or disabsorptive syndromes. Neurological features classically include sensory ataxia, impaired deep sensitivity, peripheral neuropathy and pyramidal signs. Serum levels of B12 are usually decreased, but high levels of serum homocystein and methylmalonic acid are also useful to suggest the diagnosis. In addition, MRI may show high intensity signals in the posterior column of cervical or thoracic spinal cord. Intramuscular replacement of vitamin B12 should be promptly started 3434 . Martinez Estrada KM, Cadabal Rodriguez T, Miguens Blanco I, Garcia Méndez L. Neurological signs due to isolated vitamin B12 deficiency. Semergen 2013;39:8-11. .
Vitamin E deficiency
Ataxia caused by vitamin E deficiency is an autosomal recessive disorder, and generally has a childhood onset. Rarely vitamin E deficiency causes ataxia in adulthood. The clinical spectrum resembles Friedreich ataxia: progressive ataxia, loss of proprioception, areflexia and Babinski sign 3535 . Harding AE, Muller DP, Thomas PK, Willison HJ. Spinocerebellar degeneration secondary to chronic intestinal malabsorption: a vitamin E deficiency syndrome. Ann Neurol 1982;12:419-424. . Brain MRI is usually normal, and cerebellar atrophy is uncommon. Replacement with vitamin E usually improves the symptoms 3636 . Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 2007;6:245-257. .
Multiple system atrophy - cerebellar form
Multiple system atrophy (MSA) is an adult onset sporadic neurodegenerative disease whose clinical spectrum is comprised by parkinsonian features (MSA-P) or cerebellar signs (MSA-C). In both, pyramidal signs are frequent and autonomic dysfunction is mandatory. Autonomic dysfunction is variable, and mostly related to urinary disturbances and postural hypotension. The estimated annual prevalence of the disease is 0.6:100.000 per year, reaching 3:100.000 in populations over 50 years. Median survival is 8-10 years. The underlying neuropathological findings in MSA are the presence of glial cytoplasmatic inclusions in oligodendroglial cells at autopsy. Due to the abundant presence of alpha-synuclein in this disease, similar to Parkinson's disease and dementia with Lewy bodies, MSA is currently classified as synucleinopathie. Patients with MSA present a widespread involvement of the central nervous system, with degeneration including basal ganglia, brainstem, cerebellum, pyramidal tracts and spinal cord 3737 . Stefanova N, Bücke P; Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009;8:1172-1178. .
Historically, the previous term olivopontocerebellar atrophy, when associated with dysautonomia, is in fact the so called MSA. In MSA-C, the clinical features are mainly characterized by cerebellar ataxia, ocular motility abnormalities and dysarthria. Eventually, patients with MSA-C are difficult to be distinguished from other forms of adult-onset ataxia. Autonomic dysfunction may be mild or absent in the beginning of the ataxia symptoms, and appear in the course of the disease progression. Response to levodopa is poor in MSA-C 3737 . Stefanova N, Bücke P; Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009;8:1172-1178. , 3838 . Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy—an analysis of 230 Japanese patients. Brain 2002;125:1070-1083. .
Typical neuroimaging features in MSA-C include hyperintense signal in cerebellar peduncles, olivopontocerebellar atrophy (cerebellum and brainstem), and "hot cross bun" sign, which indicates degeneration of the pons fibers ( Figure 2 ) 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. , 3737 . Stefanova N, Bücke P; Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009;8:1172-1178. .
. Brain MRI of a patient with multiple system atrophy, cerebellar form (MSA-C). Axial T1-weighted sequence: note the “hot cross bun" sign indicated by the arrow, with atrophy of the brainstem and cerebellum (A). Sagittal T1-weighted sequence: there is clear olivopontocerebellar atrophy (cerebellum and brainstem involvement) (B).
Idiopathic late-onset cerebellar ataxia
Several patients present with slow progressive pure cerebellar ataxia syndrome in adulthood, or sometimes in elderly, with no remarkable family history. Complementary investigation for sporadic ataxias is frequently inconclusive. In general, this group of patients has a degeneration restricted to the cerebellum, sparing the brainstem. This neurological condition is better known as idiopathic late-onset cerebellar ataxia (ILOCA) 3939 . Klockgether T, Ludtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain 1998;121:589-600. , 4040 . Abele M, Minnerop M, Urbach H, Specht K, Klockgether T. Sporadic adult onset ataxia of unknown etiology: a clinical, electrophysiological and imaging study. J Neurol 2007;254:1384-1389. . Substantial phenotype variability exists in ILOCA, which suggests that there are several still unknown genetic or degenerative causes. In opposite to MSA-C, brain MRI discloses atrophy limited to the cerebellum. Notably, disease progression is lower in ILOCA than in MSA-C, and life expectancy is probably normal. Some patients are mistakenly diagnosed with ILOCA, but dysautonomia may appear in the course of the disease, supporting the diagnosis of MSA-C. The diagnosis of ILOCA may be carefully done, by ruling out other acquired and genetic disease. To date, etiology and pathophysiology of ILOCA remain unclear. It is uncertain whether ILOCA is a homogeneous disease or a group of phenotypically similar syndromes represented by different neurodegenerative or genetic entities 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. , 3939 . Klockgether T, Ludtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain 1998;121:589-600. , 4040 . Abele M, Minnerop M, Urbach H, Specht K, Klockgether T. Sporadic adult onset ataxia of unknown etiology: a clinical, electrophysiological and imaging study. J Neurol 2007;254:1384-1389. .
Ataxias due to infectious diseases
Progressive adult onset sporadic ataxias may rarely be caused by chronic infections of the central nervous system. The most common infections related to progressive sporadic ataxia are neurosyphilis, Whipple's disease, Lyme disease and HIV.
The classical example of progressive ataxia caused by infection of the nervous system is neurosyphilis (bacterium Treponema pallidum ). In neurosyphilis, ataxia is purely sensory associated with pain, bladder dysfunction, and abnormal pupillary reflexes. Ataxia is due to the involvement of the posterior horn of the spinal cord 4141 . Tong ML, Lin LR, Zhang HL, et al. Spectrum and characterization of movement disorders secondary to neurosyphilis. Parkinsonism Relat Disord 2013;19:441-445. .
Whipple's disease is a systemic infection caused by the bacterium Tropheryma whipplei . The classical clinical manifestations of Whipple’s disease include fever, weight loss, diarrhea and arthritis, whereas the most common neurological manifestations are neuropsychiatric disorders, cognitive impairment and ataxia. Diagnosis is made through duodenal biopsy (PAS-positive, non-acid-fast macrophage inclusions). Blood or CSF PCR analysis may be confirmatory 4242 . Matthews BR, Jones LK, Saad DA, Aksamit AJ, Josephs KA. Arch Neurol 2005;62:618-620. .
Finally, progressive ataxia may rarely be observed in HIV-infected patients, and cerebellar syndrome occurs most commonly due to lesions of the cerebellum for opportunistic infections 4343 . Kwakwa HA, Ghobrial MW. Primary cerebellar degeneration and HIV. Arch Intern Med 2001;161:1555-1556. .
Prion diseases
Human prion diseases are a group of rare neurodegenerative disorders characterized by the conversion of the constitutively expressed prion protein, PrP(C), into an abnormally aggregated isoform, called PrP (Sc). There are four human prion diseases: Creutzfeldt-Jakob disease, Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia and Kuru. Creutzfeldt-Jakob disease, the most common disease of this group, is a rapidly progressive dementia with cortical blindness, myoclonus and cerebellar ataxia. Up to 70% of patients die within 6 months from first symptoms onset. The electroencephalogram (pseudoperiodic sharp), raised CSF 14-3-3 protein and neuron-specific enolase are helpful findings 4444 . Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224-233. . Bilateral hyperintensity of the thalamic pulvinar nucleus in T2, FLAIR and DW sequences (pulvinar sign) in brain MRI is frequently related 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. . Gerstmann-Straussler-Scheinker disease is a prionic disorder that causes ataxia and dementia. It is almost always an autosomal dominant inherited disorder and there are only few families described around the world. Onset of the disease usually occurs between the ages of 35 and 55 4444 . Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224-233. .
Other unusual sporadic ataxias
Superficial siderosis
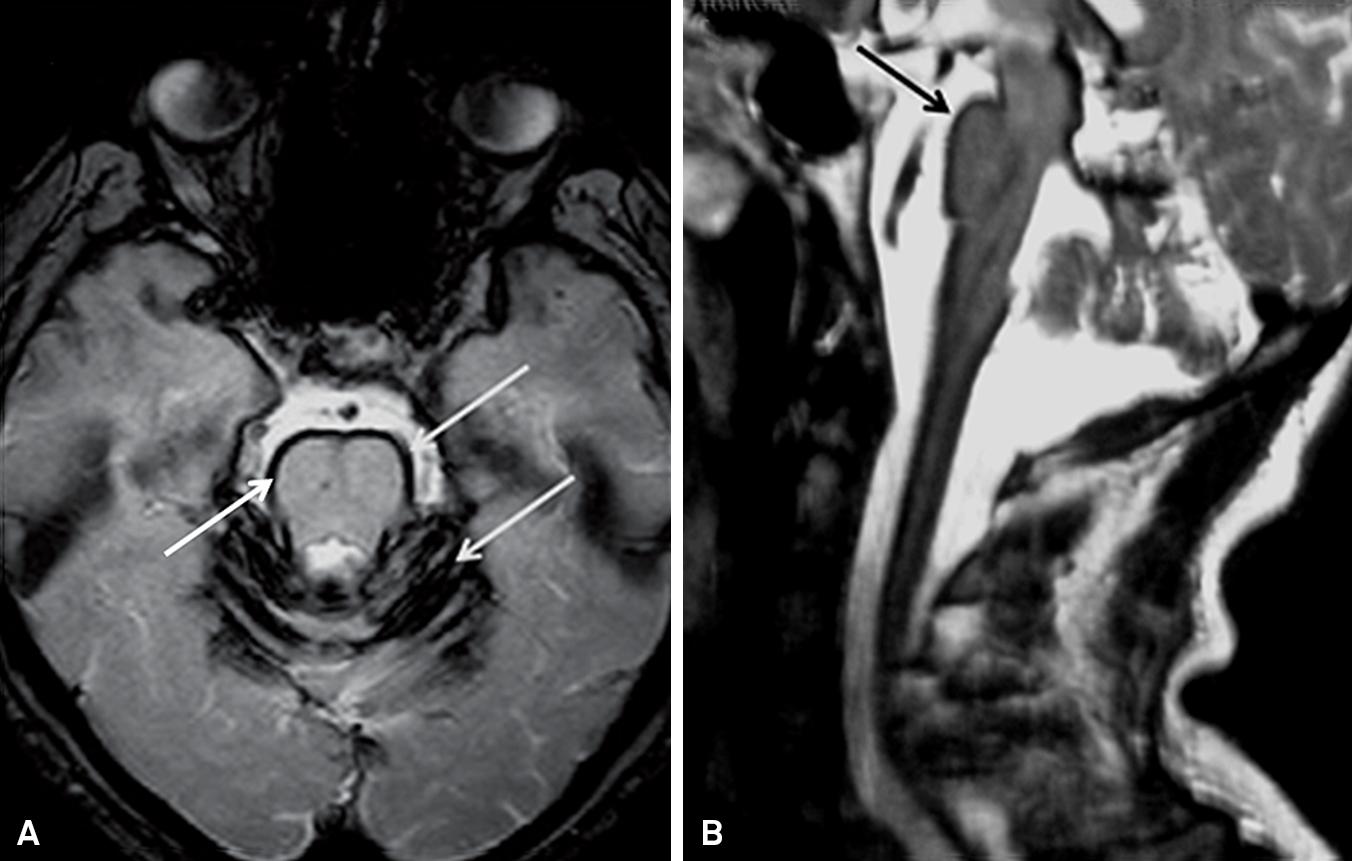
Superficial siderosis is a disorder characterized by free iron hemosiderin (a product of the breakdown of blood) deposition in the pial and subpial layer of the CNS. This condition is characterized by a triad of symptoms comprised by sensorineural hearing loss, cerebellar ataxia and pyramidal signs 4545 . Fearnley JM, Stevens JM, Rudge P. Superficial siderosis of the central nervous system. Brain 1995;118:1051-1066. . Ataxia is frequently the predominant and first symptom 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. , 4545 . Fearnley JM, Stevens JM, Rudge P. Superficial siderosis of the central nervous system. Brain 1995;118:1051-1066. . T2-weigheted brain MRI is pathognomonic, by demonstrating hypointense signal around the brainstem, cerebellum and spinal cord ( Figure 3 ). Among reported cases of secondary superficial siderosis, current or previous CNS tumors are the most common causes, followed by head or spine trauma, and arteriovenous malformations/aneurysms. However, in some cases, the source of bleeding remains obscure, despite extensive imaging and angiography. Once symptoms have appeared, the goal of therapy consists in preventing progression by ablating the origin of subarachnoid hemorrhage when correctly identified 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. , 4545 . Fearnley JM, Stevens JM, Rudge P. Superficial siderosis of the central nervous system. Brain 1995;118:1051-1066. .
. Brain MRI of a patient with superficial siderosis. Axial T2-weighted sequence: linear hypointense signal (arrow), characterizing iron deposition on the surface of the brainstem and cerebellum (A). Sagittal T2-weighted sequence demonstrating hypointense signal and iron deposition around the brainstem, and and spinal cord compression due to herniated posterior disc (postoperative) (B).
Progressive ataxia and palatal tremor
Progressive ataxia and palatal tremor (PAPT) represents a neurodegenerative disorder characterized by late-onset progressive cerebellar ataxia, oculomotor disturbances and symptomatic palatal tremor 4646 . Samuel M, Torun N, Tuite PJ, Sharpe JA, Lang AE. Progressive ataxia and palatal tremor (PAPT): clinical and MRI assessment with review of palatal tremors. Brain 2004;127:1252-1268. . Typical neuroimaging features include hypertrophy and hyperintense signal in the olivary complexes, and cerebellar atrophy 4747 . Yared JH, Lopes BS, Rogerio RM, do Amaral LL, Ferreira NF. Progressive ataxia and palatal tremor: T1-weighted with magnetization transfer pulse hyperintensity in the inferior olivary nucleus. Arq Neuropsiquiatr 2013;71:264-265. . PAPT may be divided into sporadic and familial forms. The cause of sporadic PAPT remains uncertain 4646 . Samuel M, Torun N, Tuite PJ, Sharpe JA, Lang AE. Progressive ataxia and palatal tremor (PAPT): clinical and MRI assessment with review of palatal tremors. Brain 2004;127:1252-1268. .
Histiocytosis of the nervous system
Histiocytosis, both Langerhans and non-Langerhans cell type, may be associated with cerebellar white matter changes
4848 . Candeias da Silva C, Pedroso JL, Moraes FM, et al. Teaching Neuroimages: Rosai-Dorfman disease presenting with progressive early-onset cerebellar ataxia. Neurology 2013;81:27-28.
,
4949 . Van der Knaap MS, Arts WF, Garbern JY, et al. Cerebellar leukoencephalopathy: most likely histiocytosis-related. Neurology 2008;71:1361-1367.
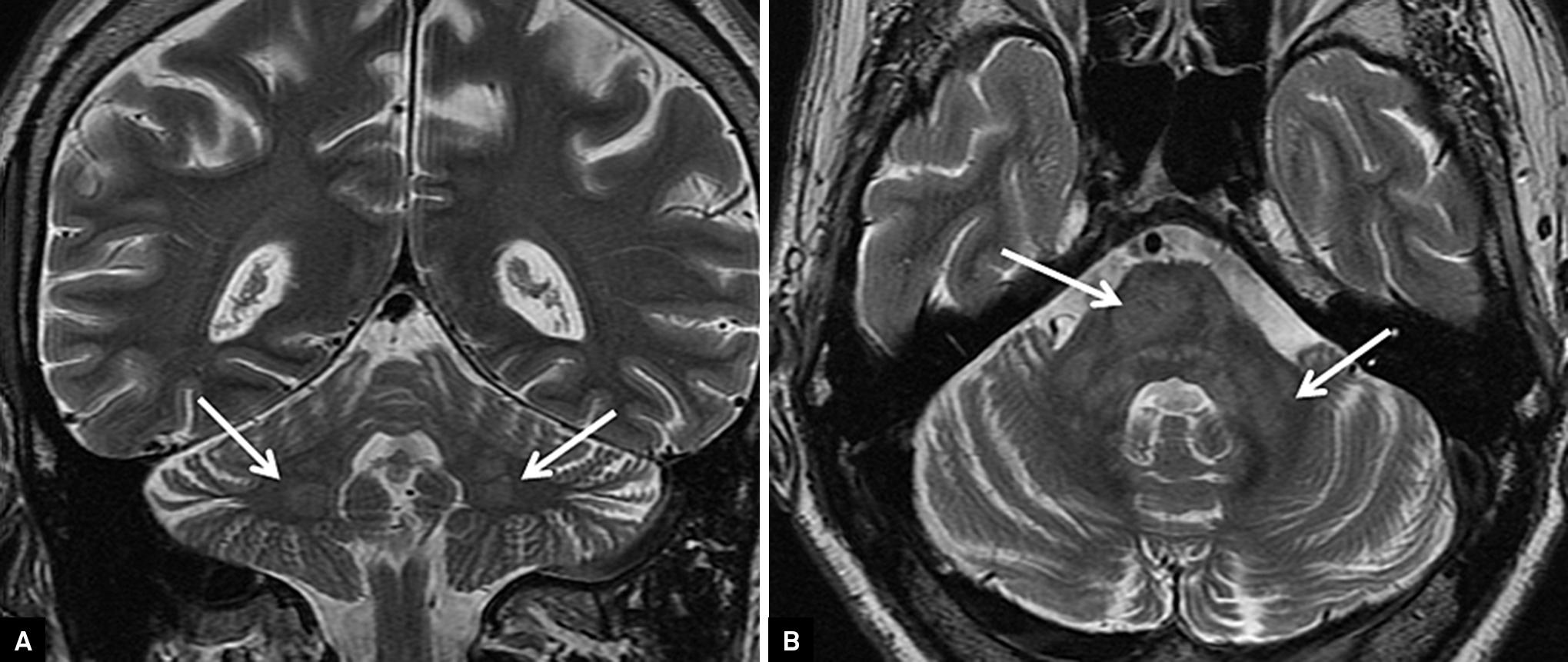
. The clinical spectrum is characterized by ataxia and pyramidal signs, and, sometimes, by cognitive dysfunction. Neuroimaging features include cerebellar white matter changes, as well as brainstem and basal ganglia abnormalities (
Figure 4
). Patients with cerebellar white matter abnormalities should be monitored for histiocytosis
4848 . Candeias da Silva C, Pedroso JL, Moraes FM, et al. Teaching Neuroimages: Rosai-Dorfman disease presenting with progressive early-onset cerebellar ataxia. Neurology 2013;81:27-28.
49 . Van der Knaap MS, Arts WF, Garbern JY, et al. Cerebellar leukoencephalopathy: most likely histiocytosis-related. Neurology 2008;71:1361-1367.
-
5050 . Van de Warrenburg BP, van der Heijden HF, Pieters G, Kremer HP. Langerhanścell histiocytosis presenting with progressive spinocerebellar ataxia. J Neurol 2003;250:1112-1114.
.
. This is a brain MRI of a patient with histiocytosis. Coronal T2-weighted brain MRI shows cerebellar white matter (dentate nucleus) involvement (arrows) (A); Axial T2-weighted brain MRI discloses hyperintense lesions in middle cerebellar peduncles and pons (arrows) (B).
Hereditary ataxias
Autosomal recessive ataxias
Although unusual, hereditary ataxias may eventually present with sporadic adult onset. Late-onset Friedreich ataxia (LOFA) has adult onset, particularly in patients with small CAG expansions
5050 . Van de Warrenburg BP, van der Heijden HF, Pieters G, Kremer HP. Langerhanścell histiocytosis presenting with progressive spinocerebellar ataxia. J Neurol 2003;250:1112-1114.
51 . Pandolfo M. Friedreich`s ataxia: clinical aspects and pathogenesis. Semin Neurol 1999;19:311-321.
-
5252 . Jiménez-Caballero PE, Marsal-Alonso C. Late onset Friedreich's ataxia. Rev Neurol 2009;48:273-274.
. Approximately 15% of patients with Friedreich ataxia have the beginning of symptoms over 25-year-old. Interestingly, there are some reports of Friedreich ataxia patients with onset of ataxia symptoms after 60-year- of-old. Usually, patients with LOFA have pyramidal signs, spasticity and brisk deep tendon reflexes, in opposite to the classical phenotype of Friedreich ataxia
5151 . Pandolfo M. Friedreich`s ataxia: clinical aspects and pathogenesis. Semin Neurol 1999;19:311-321.
,
5252 . Jiménez-Caballero PE, Marsal-Alonso C. Late onset Friedreich's ataxia. Rev Neurol 2009;48:273-274.
.
Autosomal dominant spinocerebellar ataxias
The autosomal dominant spinocerebellar ataxias (SCAs) should not be tested routinely in patients with sporadic adult-onset ataxia. However, few reports have demonstrated some SCAs with this presentation, particularly SCA6. Presumed explanations for SCAs presenting with sporadic adult onset ataxia include: de novo mutation, doubtful paternity, and small CAG expansions 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. .
Fragile-X-associated tremor/ataxia syndrome
Another hereditary ataxia with late onset and sporadic presentation is the Fragile-X-associated tremor/ataxia syndrome. This condition occurs in men (X-linked disease), although rare reports have been described in women, by mutation in the FMR1 gene and expansion of the trinucleotide CGG repeats between 55 and 200, unlike the classic fragile X syndrome, which occurs for expansion up to 200 repetitions. The clinical spectrum of this syndrome includes adult-onset ataxia associated with tremor and mild cognitive impairment 11 . Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104. , 5353 . Leehey MA, Hagerman PJ. Fragile X-associated tremor/ataxia syndrome. Handb Clin Neurol 2012;103:373-386. . Brain imaging typically discloses hyperintense signal in the dentate nucleus extending to the middle cerebellar peduncle bilaterally, as well as global changes in cerebral white matter. The diagnosis is confirmed when pre-mutation between 55 and 200 repeats in the FMR1 gene is found 5353 . Leehey MA, Hagerman PJ. Fragile X-associated tremor/ataxia syndrome. Handb Clin Neurol 2012;103:373-386. .
Adult-onset Alexander disease
Adult-onset Alexander disease is an underdiagnosed genetic entity, clinically characterized by progressive pyramidal signs, cerebellar ataxia, palatal tremor and bulbar palsy. The disease is caused by glial fibrillary acidic protein (GFAP) gene mutation. Spinal cord and medulla ablongata atrophy are typical neuroimaging features 5454 . Rezende SA, Fernandes M, Munhoz RP, et al. Cerebellar ataxia as the first manifestation of Alexander`s disease. Arq Neuropsiquiatr 2012;70:309-310. .
Mitochondrial and other genetic diseases
Mitochondrial diseases may present with adult onset ataxia, and these cases are mainly related to mutation of mitochondrial DNA polymerase γ (POLG). Furthermore, storage diseases, such as Niemann-Pick type C disease, Tay-Sachs disease, Krabbe disease, and other forms of recessive ataxias, such as ataxia with ocular apraxia and ataxia-telangiectasia may rarely present with adult onset ataxia 22 . Manto M, Marmolino D. Cerebellar ataxias. Current opinion in neurology. 2009;22:419-429. , 3636 . Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 2007;6:245-257. , 5555 . Schicks J, Müller Vom Hagen J, Bauer P, et al. Niemann-Pick disease type C is frequent in adult ataxia with cognitive decline and vertical gaze palsy. Neurology 2013;80:1169-1170. . Finally, sporadic ataxia may be the clinical presentation of new mutations recently described in autosomal recessive ataxias with adult onset of symptoms, such as SYNE1 mutations 5656 . Pedroso JL, Braga-Neto P, Ricarte IF, Albuquerque MV, Barsottini OG. Clinical spectrum of early onset cerebellar ataxia with retained tendon reflexes: na autosomal recessive ataxia not to be missed. Arq Neuropsiquiatr 2013;71:345-348. .
Final remarks
To diagnosis the exact etiology of a adult onset sporadic ataxia is a challenge. Careful attention to history, neurological examination and imaging features is crucial to guide investigations and shorten the long list of diagnostic possibilities in a patient with sporadic and non-familiar adult-onset ataxia. It is essential to target first on structural lesions (e.g. tumors, stroke and multiple sclerosis) and treatable conditions. Complementary, laboratorial and genetic investigation studies are generally excessively requested in patients with sporadic ataxia. When initial laboratorial studies do not yield a specific or obviously treatable condition in a patient with progressive sporadic ataxia, the possibility of a primary degenerative condition becomes more likely. Table summarizes the clinical spectrum, laboratory findings and neuroimaging features of the main causes of adult onset sporadic ataxias.
Enormous progress in the discoveries for genetic and molecular causes of ataxias has been made for the last decades. Furthermore, definition of clinical criteria and standard for imaging features has facilitated the correct diagnosis of the numerous forms of sporadic ataxias. Despite so many genetic discoveries, ease for imaging tests, and all laboratory investigation described in this review, etiology of sporadic ataxia remains a diagnostic challenging for the general neurologist.
References
-
1Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 2010;9:94-104.
-
2Manto M, Marmolino D. Cerebellar ataxias. Current opinion in neurology. 2009;22:419-429.
-
3Mariotti C, Fancellu R, Di Donato S. An overview of the patient with ataxia. J Neurol 2005;252:511-518.
-
4Van Gaalen J, van de Warrenburg BP. A practical approach to late-onset cerebellar ataxia: putting the disorder with lack of order into order. Pract Neurol 2012;12:14-24.
-
5Yokota O, Tsuchiya K, Terada S, et al. Frequency and clinicopathological characteristics of alcoholic cerebellar degeneration in Japan: a cross-sectional study of 1,509 postmortems. Acta Neuropathol 2006;112:43-51.
-
6Hillbom M, Muuronen A, Holm L, Hindmarsh T. The clinical versus radiological diagnosis of alcoholic cerebellar degeneration. J Neurol Sci 1986;73:45-53.
-
7Torvik A, Torp S. The prevalence of alcoholic cerebellar atrophy. A morphometric and histological study of an autopsy material. J. Neurol Sci 1986;75:43-51.
-
8Jaatinen P, Rintala J. Mechanisms of ethanol-induced degeneration in the developing, mature, and aging cerebellum. Cerebellum 2008;7:332-347.
-
9Butterworth RF. Pathophysiology of alcoholic brain damage: synergistic effects of ethanol, thiamine deficiency and alcoholic liver disease. Metab Brain Dis 1995;10:1-8.
-
10Niethammer M, Ford B. Permanent lithium-induced cerebellar toxicity: three cases and review of literature. Mov Disord 2007;22:570-573.
-
11De Marcos FA, Ghizoni E, Kobayashi E, Li LM, Cendes F. Cerebellar volume and long-term use of phenytoin. Seizure 2003;12:312-315.
-
12Orr CF, Ahlskog JE. Frequency, characteristics, and risk factors for amiodarone neurotoxicity. Arch Neurol 2009;66:865-869.
-
13Fornazzari L, Wilkinson DA, Kapur BM, Carlen PL. Cerebellar, cortical and functional impairment in toluene abusers. Acta Neurol Scand 1983;67:319-329.
-
14Bygrave HA, Geh JI, Jani Y, Glynne-Jones R. Neurological complications of 5-fluorouracil chemotherapy: case report and review of the literature. Clin Oncol (R Coll Radiol) 1998;10:334-336.
-
15Larsen JO, Braendgaard H. Structural preservation of cerebellar granule cells following neurointoxication with methyl mercury: a stereological study of the rat cerebellum. Acta Neuropathol 1995;90:251-256.
-
16Gordon MF, Abrams RI, Rubin DB, Barr WB, Correa DD. Bismuth toxicity. Neurology 1994;44:2418.
-
17Teive HA, Munhoz RP, Werneck LC. Acquired cerebellar ataxia due to statin use. Arq Neuropsiquiatr 2012;70:152.
-
18Jain KK. Drug-induced cerebellar disorders. In: Jain KK (Ed.). Drug-induced neurological disorders, 1sted. Hogrefe and Huber Publishers, Seatle, 1996:275-282.
-
19Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008;7:327-340.
-
20Scaravilli F, An SF, Groves M, Thom M. The neuropathology of paraneoplastic syndromes. Brain Pathol 1999;9:251-260.
-
21Shams’ili S, Grefkens J, de Leeuw B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain 2003;126:1409-1418.
-
22Vernino S. Paraneoplastic cerebellar degeneration. Handb Clin Neurol 2012;103:215-223.
-
23Scheid R, Voltz R, Briest S, Kluge R, von Cramon DY. Clinical insights into paraneoplastic cerebellar degeneration. J Neurol Neurosurg Psychiatry 2006;77:529-530.
-
24Aragão M de M, Pedroso JL, Albuquerque MV, Dutra LA, Barsottini OG. Superior cerebellar hyperintense sign on FLAIR-weighted magnetic resonance imaging in paraneoplastic cerebellar degeneration. Arq Neuropsiquiatr 2012;70:967.
-
25De Andrés C, Esquivel A, de Villoria JG, Graus F, Sánchez-Ramón S. Unusual magnetic resonance imaging and cerebraspinal fluid findings in paraneoplastic cerebellar degeneration: a sequential study. J Neurol Neurosurg Psychiatry 2006;77:562-563.
-
26David YB, Warner E, Levitan M, Sutton DM, Malkin MG, Dalmau JO. Autoimmune paraneoplastic cerebellar degeneration in ovarian carcinoma patients treated with plasmapheresis and immunoglobulin. A case report. Cancer 1996;78:2153-2156.
-
27Hadjivassiliou M. Immune-mediated acquired ataxias. Handb Clin Neurol 2012;103:189-199.
-
28Fernandes M, Munhoz RP, Carrilho PE, et al. Neurological disorders associated with glutamic acid decarboxylase antibodies: a Brazilian series. Arq Neuropsiquiatr 2012;70:657-661.
-
29Pedroso JL, Braga-Neto P, Dutra LA, Barsottini OG. Cerebellar ataxia associated to anti-glutamic acid decarboxylase autoantibody (anti-GAD): partial improvement with intravenous immunoglobulin therapy. Arq Neuropsiquiatr 2011;69:993.
-
30Hadjivassiliou M, Grunewald RA, Chattopadhyay AK, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet 1998;352:1582-1585.
-
31Hadjivassiliou M, Sanders DS, Woodroofe N, Williamson C, Grünewald RA. Gluten ataxia. Cerebellum 2008;7:494-498.
-
32Castillo P, Woodruff B, Caselli R, et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol 2006;63:197-202.
-
33Spinazzi M, Angelini C, Patrini C. Subacute sensory ataxia and optic neuropathy with thiamine deficiency. Nat Rev Neurol 2010;6:288-293.
-
34Martinez Estrada KM, Cadabal Rodriguez T, Miguens Blanco I, Garcia Méndez L. Neurological signs due to isolated vitamin B12 deficiency. Semergen 2013;39:8-11.
-
35Harding AE, Muller DP, Thomas PK, Willison HJ. Spinocerebellar degeneration secondary to chronic intestinal malabsorption: a vitamin E deficiency syndrome. Ann Neurol 1982;12:419-424.
-
36Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 2007;6:245-257.
-
37Stefanova N, Bücke P; Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009;8:1172-1178.
-
38Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy—an analysis of 230 Japanese patients. Brain 2002;125:1070-1083.
-
39Klockgether T, Ludtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain 1998;121:589-600.
-
40Abele M, Minnerop M, Urbach H, Specht K, Klockgether T. Sporadic adult onset ataxia of unknown etiology: a clinical, electrophysiological and imaging study. J Neurol 2007;254:1384-1389.
-
41Tong ML, Lin LR, Zhang HL, et al. Spectrum and characterization of movement disorders secondary to neurosyphilis. Parkinsonism Relat Disord 2013;19:441-445.
-
42Matthews BR, Jones LK, Saad DA, Aksamit AJ, Josephs KA. Arch Neurol 2005;62:618-620.
-
43Kwakwa HA, Ghobrial MW. Primary cerebellar degeneration and HIV. Arch Intern Med 2001;161:1555-1556.
-
44Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224-233.
-
45Fearnley JM, Stevens JM, Rudge P. Superficial siderosis of the central nervous system. Brain 1995;118:1051-1066.
-
46Samuel M, Torun N, Tuite PJ, Sharpe JA, Lang AE. Progressive ataxia and palatal tremor (PAPT): clinical and MRI assessment with review of palatal tremors. Brain 2004;127:1252-1268.
-
47Yared JH, Lopes BS, Rogerio RM, do Amaral LL, Ferreira NF. Progressive ataxia and palatal tremor: T1-weighted with magnetization transfer pulse hyperintensity in the inferior olivary nucleus. Arq Neuropsiquiatr 2013;71:264-265.
-
48Candeias da Silva C, Pedroso JL, Moraes FM, et al. Teaching Neuroimages: Rosai-Dorfman disease presenting with progressive early-onset cerebellar ataxia. Neurology 2013;81:27-28.
-
49Van der Knaap MS, Arts WF, Garbern JY, et al. Cerebellar leukoencephalopathy: most likely histiocytosis-related. Neurology 2008;71:1361-1367.
-
50Van de Warrenburg BP, van der Heijden HF, Pieters G, Kremer HP. Langerhanścell histiocytosis presenting with progressive spinocerebellar ataxia. J Neurol 2003;250:1112-1114.
-
51Pandolfo M. Friedreich`s ataxia: clinical aspects and pathogenesis. Semin Neurol 1999;19:311-321.
-
52Jiménez-Caballero PE, Marsal-Alonso C. Late onset Friedreich's ataxia. Rev Neurol 2009;48:273-274.
-
53Leehey MA, Hagerman PJ. Fragile X-associated tremor/ataxia syndrome. Handb Clin Neurol 2012;103:373-386.
-
54Rezende SA, Fernandes M, Munhoz RP, et al. Cerebellar ataxia as the first manifestation of Alexander`s disease. Arq Neuropsiquiatr 2012;70:309-310.
-
55Schicks J, Müller Vom Hagen J, Bauer P, et al. Niemann-Pick disease type C is frequent in adult ataxia with cognitive decline and vertical gaze palsy. Neurology 2013;80:1169-1170.
-
56Pedroso JL, Braga-Neto P, Ricarte IF, Albuquerque MV, Barsottini OG. Clinical spectrum of early onset cerebellar ataxia with retained tendon reflexes: na autosomal recessive ataxia not to be missed. Arq Neuropsiquiatr 2013;71:345-348.
Publication Dates
-
Publication in this collection
Mar 2014
History
-
Received
24 Aug 2013 -
Reviewed
01 Nov 2013 -
Accepted
08 Nov 2013