ABSTRACT
We describe a six-year-old boy with a history of hematuria, posterior polymorphous corneal dystrophy and dots and fleck retinopathy. Alport syndrome should be ruled out in patients presenting with posterior polymorphous corneal dystrophy or anterior lenticonus and a family history of renal disease.
Keywords:
Corneal dystrophy; hereditary/etiology; Retinal diseases/etiology; Lens diseases/etiology; Nephritis, hereditary/complications; Hematuria
RESUMO
Descrevemos um paciente de 6 anos de idade com história de hematúria, distrofia corneana polimorfa posterior e retinopatia em "dots and flecks". Síndrome de Alport deve ser excluída se o paciente apresentar com distrofia corneana polimorfa posterior ou lenticone anterior com historia familiar de doença renal.
Descritores:
Distrofias hereditárias da córnea; Doenças retinianas/etiologia; Doenças do cristalino/etiologia; Nefrite hereditária/complicações; Hematúria
INTRODUCTION
X-linked Alport Syndrome is commonly associated with retinal dots and flecks as well as anterior lenticonus11 Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet. 1997; 18(4):161-73.. Conversely, posterior polymorphous corneal dystrophy is a rare finding in this disorder. Boys with this corneal finding should be screened for hearing impairment and renal disease even in the absence of anterior lenticonus.
CASES REPORT
A 6 year old male with persistent proteinuria and gross hematuria was referred by Paediatric Nephrology for ophthalmic examination with suspected Alport syndrome. His mother found him to have gross hematuria at the age of 3 years. He had also been tested for hearing changes annually and had normal hearing so far.
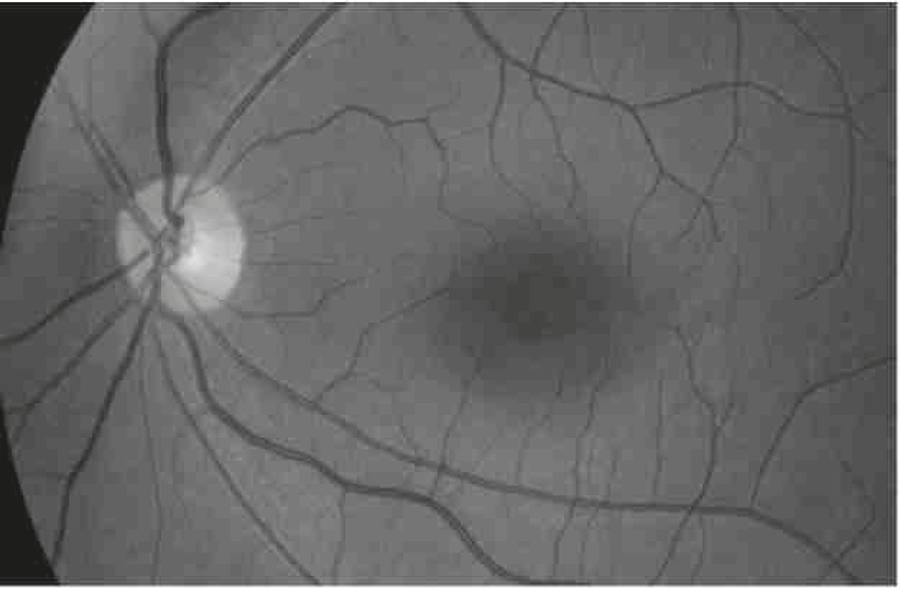
At the time of eye exam he was found to have normal visual acuities in both eyes with good stereopsis. On slit lamp examination he had left eye posterior polymorphous dystrophy (Figure 1). Both lenses were clear with no sign of anterior lenticonus. Fundoscopy showed healthy optic discs in both eyes and maculae with broadening of fovea reflex bilaterally with yellowish flecks (Figure 2).
Posterior polymorphous corneal dystrophy. The 'snail track' endothelial appearance is evident on this slit-lamp photograph
His mother had a history of an episode of hematuria at the age of 12 years. His aunt and cousin also had a positive history of hematuria. His paternal grandfather died at the age of 25 years of kidney disease and his maternal uncle died at the age of 40 of unknown causes. His maternal great grand father had also died of kidney disease at the age of 30. The patient's brother also had hematuria and was seen in the eye clinic with a normal eye examination.
Genetic test for COL4A5 gene was done to confirm the boys' diagnosis of X-linked Alport syndrome. Sequencing showed a mutation that changes glycine residue: c.3544G>A (p.G1182S) A Glycine residue must be present at every third position to allow proper folding of the collagen protein. Both boys have been having annual follow-up and advised to continue enalapril as prescribed by the nephrology clinic.
DISCUSSION
Alport syndrome is a rare genetic disorder which affects 1 in 50,000 live births22 Mukerji N, Dodson K. X-linked Alport syndrome: a case report. Internet J Nephrol. 2003;1:42. [cited 2015 Jul 26]. Available from: http://ispub.com/IJNE/1/1/1339
http://ispub.com/IJNE/1/1/1339...
.
The inheritance is predominantly X-linked (85%) but may be autosomal recessive (10%) or autosomal dominant (5%). Men are typically severely affected, and female carriers tend to have a mild form of the disease, often microscopic hematuria with normal renal function. 33 Seymenoðlu G, Baser EF. Ocular manifestations and surgical results in patients with Alport syndrome. J Cataract Refract Surg. 2009;35(7):1302-6. It is primarily a basement membrane disorder caused by mutations in COL4A3, COL4A4 and COL4A5 collagen biosynthesis genes44 Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotypephenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol.2003 Oct;14(10):2603-10.. These mutations prevent the formation of Type IV collagen fibres. The mutations demonstrated in X-Linked Alport syndrome affect the COLqAg gene which encodes the alpha 5 chain of Type IV collagen and as this protein is common to the basement membranes of glomerulus, cornea, lens capsule, retina and cochlear44 Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotypephenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol.2003 Oct;14(10):2603-10. hence the manifestations in kidneys, eyes and ears in this syndrome. Furthermore, the alpha 3(IV), 4(IV) and 5(IV) molecules interfere with the stability of all three and the loss of these collagen molecules from the affected basement membrane results in the anomalous structural changes found in the eyes11 Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet. 1997; 18(4):161-73..
Eighty five percent of the affected adult males present with dot-and-fleck retinopathy and the rest of the patients present with anterior lenticonus11 Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet. 1997; 18(4):161-73.. These changes worsen with progression of disease and are frequently detected at the onset of renal failure. Posterior polymorphous corneal dystrophy which was found in our patient is a relatively rare finding. although the presence of anterior lenticonus or posterior polymorphous corneal dystrophy in a person with a family history of Alport syndrome or with endstage renal disease is a diagnostic of Alport syndrome(1, 5). Other ocular abnormalities that may be detected in Alport syndrome are poor macular reflex, microcornea, spontaneous lens rupture, spherophakia, iris atrophy, cataracts, and retinal pigmentations11 Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet. 1997; 18(4):161-73..
The primary renal manifestation is typically asymptomatic microhematuria and recurrent gross hematuria in early childhood is common, as in this clinical case.
There is no known cure for the condition. The treatment is therefore symptomatic, aiming to keep the blood pressure under control and counselling the patients to keep a watch for signs of renal failure. Once kidney failure develops, patients are offered dialysis or renal transplant, although the body may reject the new kidney as it contains normal type IV collagen which is recognised as foreign by the immune system.
-
Institution: University of British Columbia, Children's Hospital - Vancouver, BC, Canada.
REFERENCES
-
1Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet. 1997; 18(4):161-73.
-
2Mukerji N, Dodson K. X-linked Alport syndrome: a case report. Internet J Nephrol. 2003;1:42. [cited 2015 Jul 26]. Available from: http://ispub.com/IJNE/1/1/1339
» http://ispub.com/IJNE/1/1/1339 -
3Seymenoðlu G, Baser EF. Ocular manifestations and surgical results in patients with Alport syndrome. J Cataract Refract Surg. 2009;35(7):1302-6.
-
4Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotypephenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol.2003 Oct;14(10):2603-10.
-
5Kruegel J, Rubel D, Gross O. Alport syndrome-insights from basic and clinicalresearch. Nat Rev Nephrol. 2013;9(3):170-8. Review.
Publication Dates
-
Publication in this collection
Sep-Oct 2016
History
-
Received
16 Feb 2015 -
Accepted
22 Sept 2015