Abstracts
Schinzel-Giedion syndrome is a rare autosomal recessive disorder characterized by coarse facies, midface retraction, hypertrichosis, multiple skeletal anomalies, and cardiac and renal malformations. Craniofacial abnormalities of this syndrome sometimes resemble a storage or metabolic disease. The pathogenesis of the disease remains unknown. The objective of this report was to emphasize the importance of congenital bilateral hydronephrosis for the diagnosis of Schinzel-Giedion syndrome. We describe the first Brazilian case of a newborn with typical facies, generalized hypertrichosis, cardiac and skeletal anomalies, and bilateral hydronephrosis detected during pregnancy and confirmed later by abdominal ultrasonography. Chromosomal constitution was normal. Of the 35 cases already reported in the literature, 31 presented hydronephrosis, which is considered an important clue in diagnosis. If Schinzel-Giedion syndrome were indexed as a cause of congenital hydronephrosis, its identification would be greatly facilitated, since the majority of the other findings in Schinzel-Giedion syndrome are nonspecific and common to many genetic syndromes.
Schinzel-Giedion syndrome; Hydronephrosis; Midface hypoplasia
A síndrome de Schinzel-Giedion é uma patologia genética rara de etiologia desconhecida e herança autossômica recessiva. Caracteriza-se pela presença de um fácies grotesco, hipoplasia da porção média da face, hipertricose, múltiplas anomalias esqueléticas, malformações cardíacas e renais.As anomalias craniofaciais desta síndrome podem lembrar o fácies de uma doença metabólica de depósito. O objetivo deste relato foi enfatizar a importância da hidronefrose congênita bilateral no diagnóstico da síndrome de Schinzel-Giedion . Descrevemos o primeiro caso brasileiro de um recém-nascido com fácies típico, hipertricose generalizada, anomalias esqueléticas, cardíacas e hidronefrose bilateral, detectada pela ultrassonografia fetal e, posteriormente, confirmada pelo mesmo método. O estudo cromosômico foi normal. Na literatura, de 35 casos descritos, 31 apresentavam hidronefrose, o que constitui um achado fundamental para o diagnóstico da patologia. Dessa forma, acreditamos que se a síndrome de Schinzel-Giedion fosse indexada como uma das causas de hidronefrose congênita, seu diagnóstico seria facilitado, uma vez que a maioria dos outros achados desta síndrome, com exceção da hidronefrose, é inespecífica e comum a diversas outras síndromes genéticas.
Síndrome de Schinzel-Giedion; Hidronefrose; Hipoplasia da face média
CASE REPORT
Hydronephrosis in Schinzel-Giedion syndrome: an important clue for the diagnosis
Hidronefrose na síndrome de Schinzel-Giedion: um achado importante para o diagnóstico
Lilian Maria José Albano; Paula Priscila Ohara Sakae; Marta Maria Galli Bozzo Mataloun; Clea Rodrigues Leone; Débora R. Bertola; Chong Ae Kim
Clinical Genetic Unit, Children's Institute, Hospital das Clínicas, Faculty of Medicine, University of São Paulo – São Paulo/SP, Brazil
ABSTRACT
Schinzel-Giedion syndrome is a rare autosomal recessive disorder characterized by coarse facies, midface retraction, hypertrichosis, multiple skeletal anomalies, and cardiac and renal malformations. Craniofacial abnormalities of this syndrome sometimes resemble a storage or metabolic disease. The pathogenesis of the disease remains unknown.
The objective of this report was to emphasize the importance of congenital bilateral hydronephrosis for the diagnosis of Schinzel-Giedion syndrome.
We describe the first Brazilian case of a newborn with typical facies, generalized hypertrichosis, cardiac and skeletal anomalies, and bilateral hydronephrosis detected during pregnancy and confirmed later by abdominal ultrasonography. Chromosomal constitution was normal.
Of the 35 cases already reported in the literature, 31 presented hydronephrosis, which is considered an important clue in diagnosis. If Schinzel-Giedion syndrome were indexed as a cause of congenital hydronephrosis, its identification would be greatly facilitated, since the majority of the other findings in Schinzel-Giedion syndrome are nonspecific and common to many genetic syndromes.
Key words: Schinzel-Giedion syndrome. Hydronephrosis. Midface hypoplasia.
RESUMO
A síndrome de Schinzel-Giedion é uma patologia genética rara de etiologia desconhecida e herança autossômica recessiva. Caracteriza-se pela presença de um fácies grotesco, hipoplasia da porção média da face, hipertricose, múltiplas anomalias esqueléticas, malformações cardíacas e renais.As anomalias craniofaciais desta síndrome podem lembrar o fácies de uma doença metabólica de depósito.
O objetivo deste relato foi enfatizar a importância da hidronefrose congênita bilateral no diagnóstico da síndrome de Schinzel-Giedion .
Descrevemos o primeiro caso brasileiro de um recém-nascido com fácies típico, hipertricose generalizada, anomalias esqueléticas, cardíacas e hidronefrose bilateral, detectada pela ultrassonografia fetal e, posteriormente, confirmada pelo mesmo método. O estudo cromosômico foi normal.
Na literatura, de 35 casos descritos, 31 apresentavam hidronefrose, o que constitui um achado fundamental para o diagnóstico da patologia. Dessa forma, acreditamos que se a síndrome de Schinzel-Giedion fosse indexada como uma das causas de hidronefrose congênita, seu diagnóstico seria facilitado, uma vez que a maioria dos outros achados desta síndrome, com exceção da hidronefrose, é inespecífica e comum a diversas outras síndromes genéticas.
Unitermos: Síndrome de Schinzel-Giedion. Hidronefrose. Hipoplasia da face média.
Schinzel-Giedion syndrome (SGS), first described in 1978, is a rare syndrome characterized by midface retraction, hypertrichosis, multiple skeletal anomalies, and cardiac and renal malformations.
Some authors believe that if the syndrome identification were indexed as a cause of congenital hydronephrosis, its diagnosis would be considerably facilitated1. In order to reinforce this opinion and the phenotypic spectrum of the syndrome, we resolved to report another case: the first Brazilian SGS patient. To date, more than 30 cases have been reported in the literature.
CLINICAL REPORT
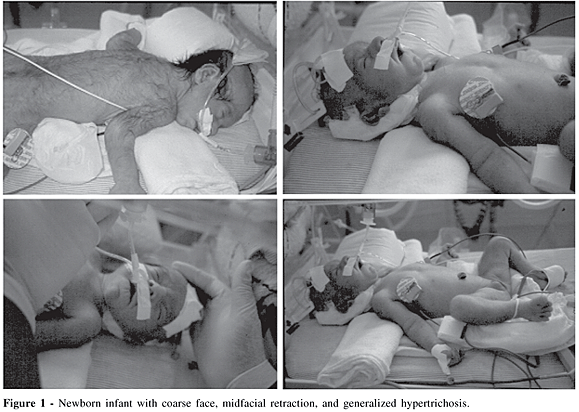
We report a newborn female infant with bilateral hydronephrosis (Fig.1); the mother was a 29-year-old woman. There was no parental consanguinity nor family history of congenital abnormalities. The baby was born at term with a birth weight = 2.440 g (10th centile); length = 48 cm (50th centile), O.F.C.= 31 (<2.5 centile); inner canthal distance = 2.3 cm (75th centile); interpupillary distance = 4.3 cm (50th centile); outer canthal distance = 5.8 cm (25th centile). Bilateral hydronephrosis was detected during pregnancy by ultrasonography. She presented generalized hypertrichosis, coarse facies with prominent forehead, widely patent fontanels and sutures, short and "squared" nose with anteverted nares and depressed bridge, ears apparently low-set and posteriorly rotated with folded helices, prominent eyes with a deep groove underneath, thin lips, supernumerary nipples, "apparent" shortening of the arms with an accessory crease, bilateral simian palmar creases, large thumbs and great toes (laterally deviated), hypoplastic nails especially in the fingers, and an anteriorly placed anus. Cardiac evaluation revealed interatrial communication, patent ductus arteriosus, pulmonary stenosis and atresia, hypoplasia of the right ventricle, and moderate mitral valve insufficiency. Abdominal ultrasonography confirmed the bilateral hydronephrosis detected in utero (Fig.2). In the x-ray skeletal survey, we noticed wide cranial sutures, sclerosis of the basal and mid-fossa brain bones, wormian occipital bones, and wide occipital synchondrosis. The ribs, clavicles, pelvis, vertebral column, and long bones were unremarkable. Centers of ossification in the knee were not observed. Chromosomal analysis was normal (Giemsa banding). The clinical course was complicated by pulmonary hypertension and heart failure, and the patient died at the age of 3 months. Unfortunately, an autopsy could not be performed.
DISCUSSION
The phenotypic characterization of SGS includes a coarse midface retraction, a prominent forehead, and an enlarged and protuberant tongue. These craniofacial abnormalities sometimes resemble a storage or metabolic disease, but patients with SGS do not have a biochemical abnormality1,2. Hirsutism disappears and midface retraction becomes less evident with age; In contrast, bitemporal narrowing becomes more evident3. In addition, radiological findings with a specific skeletal dysplasia and the presence of bilateral hydronephrosis strengthened the diagnosis of SGS.
Hydronephrosis is only occasionally reported as a feature of a malformation syndrome, such as Johansson-Blizzard syndrome, trisomy 13 and 18, Turner syndrome, triploidy, and Ochoa syndrome1.
Of the 35 SGS cases reviewed by Touge et al. (2001)4, 31 presented hydronephrosis, which is an important clue in diagnosis, and Minn et al. (2002)5 have the same opinion. Kelley et al. (1982) considered that the syndrome identification would be greatly facilitated if the Schinzel-Giedion syndrome were "indexed" as a cause of congenital hydronephrosis.
We also observed the renal anomaly in our patient and agree with the authors that the majority of the findings of this syndrome except hydronephrosis are nonspecific and common to many genetic syndromes (Table 1).
Since the gene of the disease has not yet been identified and diagnosis is strictly based on clinical findings, the presence of hydronephrosis assumes an important role for the diagnosis of SGS. Despite the lack of identification of any biochemical abnormality so far, Shah et al. (1999)6 suggested that the progressive neurodegenerative process in SGS associated with a coarse facial appearance and skeletal abnormalities could be associated with a metabolic defect.
Some authors have described sacral tumors associated with this syndrome, and McPherson et al. (1998)7 have considered that this finding could help in explaining the pathogenesis and/or identifying candidate genes for this autosomal recessive condition.
Therefore, additional patients should be reported in order to amplify the phenotypic spectrum of SGS.
Received for publication on July 08, 2003.
-
1Kelley RI, Zackai EH, Charney EB. Congenital hydronephrosis, skeletal dysplasia, and severe developmental retardation: the Schinzel-Giedion syndrome. J Pediatr 1982; 100:943-946.
-
2Al-Gazali LI, Farndon P, Burn J, et al. The Schinzel-Giedion syndrome. J Med Genet 1990; 27:42-47.
-
3Rodríguez JI, Jiménez-Heffernan JÁ, Leal J. Schinzel-Giedion syndrome: autopsy report and additional clinical manifestations. Am J Med Genet 1994; 53:374-377.
-
4Minn D, Christmann D, De Saint-Martin A, et al. Further clinical and sensorial delineation of Schinzel-Giedion Syndrome: report of two cases. Am J Med Genet 2002; 109:211-217.
-
5Touge H, Fujinaga T, Okuda M, et al. Schinzel-Giedion syndrome. Intern J Urol 2002; 8:237-241.
-
6Shah AM, Smith MF, Griffiths PD, et al. Schinzel-Giedion syndrome: evidence for a neurodegenerative process. Am J Med Genet 1999; 82: 344-347.
-
7Mcpherson E, Clemens M, Hoffner L. Sacral tumors in Schinzel-Giedion syndrome. Am J Med Genet 1998; 79: 62-63.
-
8Labrune P, Lyonnet S, Zupan V, et al. Three new cases of the Schinzel-Giedion syndrome and review of the literature. Am J Med Genet 1994; 50:90-93.
-
9Antich J, Manzanares R, Camarasa F, et al. Schinzel-Giedion syndrome: report of two sibs. Am J Med Genet 1995; 59:96-99.
-
10Özkinay FF, Akisü M, Kültürsay N, et al. Agenesis of the corpus callosum in Schinzel-Giedion syndrome associated with 47,XXY karyotype. Clin Genet 1996; 50:145-148.
-
11Shelagh J, Dean JCS. A Schinzel-Giedion-like syndrome _ a milder version or a separate condition? Clin Dysmorphol 2002: 11:271-275.
Publication Dates
-
Publication in this collection
23 Apr 2004 -
Date of issue
2004
History
-
Received
08 July 2003