Resumos
Apresentamos um caso de uma criança com dois anos e oito meses de idade,acometida pela forma recessiva da displasia craniometafisária. São enfatizados os achados clínico-radiológicos, bem como seus principais diagnósticos diferenciais, baseados em dados de breve revisão literária.
Displasia craniometafisária; Displasia óssea; Esclerose óssea; Obstrução nasal
We report a case of a 2 years and 8 months-old child with the recessive form of craniometaphyseal dysplasia. Clinical and radiological features as well as the main differential diagnosis are discussed and compared with data from a brief literature review.
Craniometaphyseal dysplasia; Bone dysplasia; Bone sclerosis; Nasal obstruction
Relato de Caso
DISPLASIA CRANIOMETAFISÁRIA - RELATO DE CASO* * Trabalho realizado no Serviço de Diagnóstico por Imagem do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO. 1. Médicos Residentes do Serviço de Diagnóstico por Imagem do HC-FMUFG. 2. Médica Radiologista Assistente do Serviço de Diagnóstico por Imagem do HC-FMUFG. 3. Professor Adjunto Doutor do Departamento de Diagnóstico por Imagem e Anatomia Patológica da FMUFG. Endereço para correspondência: Dr. Sizenildo da Silva Figueirêdo. Rua 236, nº 121, apto. 106, Setor Universitário. Goiânia, GO, 74120-110. E-mail: size75@ig.com.br Recebido para publicação em 10/1/2002. Aceito, após revisão, em 27/3/2002.
Sizenildo da Silva Figueirêdo1 * Trabalho realizado no Serviço de Diagnóstico por Imagem do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO. 1. Médicos Residentes do Serviço de Diagnóstico por Imagem do HC-FMUFG. 2. Médica Radiologista Assistente do Serviço de Diagnóstico por Imagem do HC-FMUFG. 3. Professor Adjunto Doutor do Departamento de Diagnóstico por Imagem e Anatomia Patológica da FMUFG. Endereço para correspondência: Dr. Sizenildo da Silva Figueirêdo. Rua 236, nº 121, apto. 106, Setor Universitário. Goiânia, GO, 74120-110. E-mail: size75@ig.com.br Recebido para publicação em 10/1/2002. Aceito, após revisão, em 27/3/2002. , Luiza Helena Vilela Ribeiro2 * Trabalho realizado no Serviço de Diagnóstico por Imagem do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO. 1. Médicos Residentes do Serviço de Diagnóstico por Imagem do HC-FMUFG. 2. Médica Radiologista Assistente do Serviço de Diagnóstico por Imagem do HC-FMUFG. 3. Professor Adjunto Doutor do Departamento de Diagnóstico por Imagem e Anatomia Patológica da FMUFG. Endereço para correspondência: Dr. Sizenildo da Silva Figueirêdo. Rua 236, nº 121, apto. 106, Setor Universitário. Goiânia, GO, 74120-110. E-mail: size75@ig.com.br Recebido para publicação em 10/1/2002. Aceito, após revisão, em 27/3/2002. , Bruno Barcelos da Nóbrega1 * Trabalho realizado no Serviço de Diagnóstico por Imagem do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO. 1. Médicos Residentes do Serviço de Diagnóstico por Imagem do HC-FMUFG. 2. Médica Radiologista Assistente do Serviço de Diagnóstico por Imagem do HC-FMUFG. 3. Professor Adjunto Doutor do Departamento de Diagnóstico por Imagem e Anatomia Patológica da FMUFG. Endereço para correspondência: Dr. Sizenildo da Silva Figueirêdo. Rua 236, nº 121, apto. 106, Setor Universitário. Goiânia, GO, 74120-110. E-mail: size75@ig.com.br Recebido para publicação em 10/1/2002. Aceito, após revisão, em 27/3/2002. , Kim-Ir-Sen Santos Teixeira3 * Trabalho realizado no Serviço de Diagnóstico por Imagem do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO. 1. Médicos Residentes do Serviço de Diagnóstico por Imagem do HC-FMUFG. 2. Médica Radiologista Assistente do Serviço de Diagnóstico por Imagem do HC-FMUFG. 3. Professor Adjunto Doutor do Departamento de Diagnóstico por Imagem e Anatomia Patológica da FMUFG. Endereço para correspondência: Dr. Sizenildo da Silva Figueirêdo. Rua 236, nº 121, apto. 106, Setor Universitário. Goiânia, GO, 74120-110. E-mail: size75@ig.com.br Recebido para publicação em 10/1/2002. Aceito, após revisão, em 27/3/2002.
Resumo
Apresentamos um caso de uma criança com dois anos e oito meses de idade,acometida pela forma recessiva da displasia craniometafisária. São enfatizados os achados clínico-radiológicos, bem como seus principais diagnósticos diferenciais, baseados em dados de breve revisão literária.
Unitermos: Displasia craniometafisária. Displasia óssea. Esclerose óssea. Obstrução nasal.
Abstract
Craniometaphyseal dysplasia - case report.
We report a case of a 2 years and 8 months-old child with the recessive form of craniometaphyseal dysplasia. Clinical and radiological features as well as the main differential diagnosis are discussed and compared with data from a brief literature review.
Key words: Craniometaphyseal dysplasia. Bone dysplasia. Bone sclerosis. Nasal obstruction.
INTRODUÇÃO
Displasia craniometafisária (DCM) é uma expressão criada por Jackson et al.(1), em 1954, para uma doença óssea hereditária caracterizada por alargamento metafisário dos ossos tubulares associado a esclerose e hiperostose craniofaciais (leontíase óssea)(2). A presença de proeminentes anormalidades cranianas em dois de cinco casos descritos por esses autores os fizeram acreditar que se tratava de uma entidade distinta da doença de Pyle (displasia metafisária).
A DCM representa uma desordem geneticamente heterogênea, apresentando transmissão autossômica dominante na maioria dos casos, embora a forma recessiva tenha sido observada(3-7). Há poucos relatos de herança autossômica recessiva(3,5,6-8) e esta comumente apresenta achados clínico-radiológicos mais severos(6,7,9-11). Os sintomas iniciais freqüentemente decorrem da obstrução nasal gradativa e distorção facial(7), bem como de alterações nos ossos temporais, geralmente levando a um tipo misto de surdez progressiva(6).
Devido à escassez de casos publicados, há alguma confusão em relação às características da doença(9), porém os mais significativos padrões clínicos e radiográficos estão bem fundamentados. Segundo Taybi e Lachman(12), apenas cerca de 85 casos haviam sido descritos até 1994. Relatamos o caso de um infante provavelmente acometido pela forma recessiva da DCM, corretamente diagnosticada aos dois anos e oito meses de idade.
RELATO DO CASO
Criança do sexo masculino, dois anos e oito meses de idade, foi encaminhada ao Departamento de Diagnóstico por Imagem e Anatomia Patológica do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás para investigação, devido a distorção craniofacial grosseira vista em uma radiografia de cavum realizada para avaliação de alterações tonsilares clinicamente evidentes.
Havia antecedente de cirurgia de derivação ventrículo-peritoneal (devido a hidrocefalia) realizada em outra instituição aos seis meses de idade, onde firmou-se um diagnóstico inicial de "displasia fibrosa craniofacial". Apresentava história de deformidade facial progressiva, obstrução nasal persistente, dificuldades alimentares, infecções recorrentes de vias respiratórias superiores, roncos, respiração bucal e aparente diminuição da acuidade visual. Os pais não apresentavam anormalidades fenotípicas.
Ao exame clínico evidenciamos deformidade facial caracterizada por típico hipertelorismo, prognatismo, proeminência paranasal e da raiz do nariz (násio), boca constantemente semiaberta, dentição defeituosa, tonsilas palatinas aumentadas e hiperemiadas. Circunferência craniana acima do percentil 90.
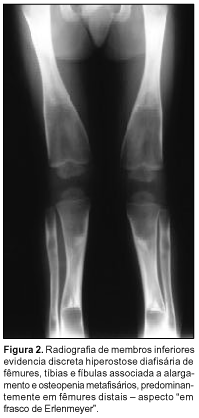
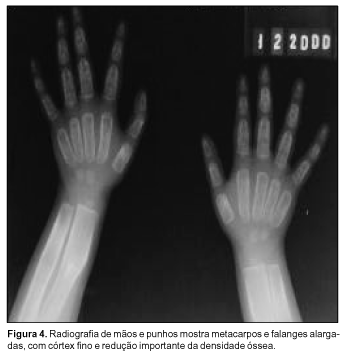
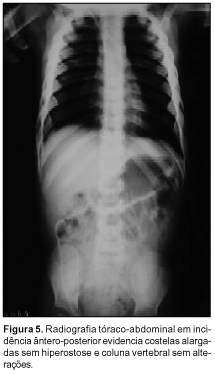
À avaliação radiográfica observamos: severa hiperostose dos ossos craniofaciais; obliteração de todos os seios paranasais e das células mastóideas; hipertelorismo; prognatismo com atenuação do ângulo mandibular (Figura 1); discreto afilamento cortical e leve esclerose diafisária associados a marcantes alargamento e radiotransparência metafisários dos ossos longos, menos proeminente nos membros superiores (Figuras 2 e 3); alargamento dos ossos metacarpofalangeanos, com córtex fino e densidade óssea diminuída (Figura 4); expansão generalizada das costelas, sem esclerose (Figura 5). Coluna vertebral, clavículas, escápulas e pelve sem alterações.
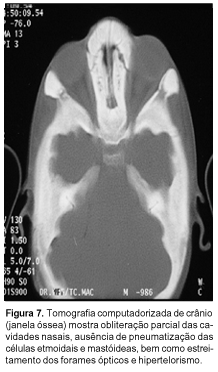
A tomografia computadorizada de crânio ratificou os achados das radiografias e delineou melhor o comprometimento dos forames dos nervos ópticos (Figuras 6 e 7).
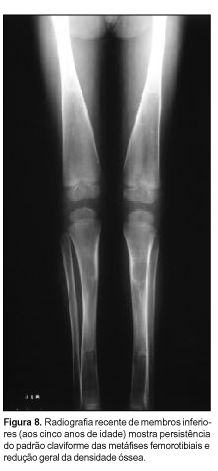
Atualmente o paciente tem cinco anos de idade, tendo evoluído com piora da acuidade visual e da deformidade facial, porém sem queixas auditivas até o momento. À avaliação oftalmológica observou-se atrofia óptica bilateral. A tomografia computadorizada para estudo de órbitas mostrou importante redução do diâmetro dos forames ópticos. Radiografias recentes de membros inferiores revelam redução da radiopacidade diafisária inicialmente presente e persistência do alargamento metafisário (Figura 8). Crânio aparentemente mais esclerótico do que nos exames prévios.
Investigação radiológica recentemente realizada em seu irmão de um ano de idade não mostrou alterações.
DISCUSSÃO
A DCM faz parte do espectro das chamadas "displasias craniotubulares", criteriosamente divididas por Gorlin et al.(13). Apesar de a determinação genética ter sido bem evidenciada, a exata patogênese não é conhecida(2,9). Talvez as alterações sejam resultado de uma falha na reabsorção (osteoclastos hipoativos) e/ou na formação ósseas (osteoblastos hiperativos)(2,14).
Os achados clínicos mais evidentes incluem: a) caracteres craniofaciais anormais - cabeça larga e grande, bossa frontal, hipertelorismo, alargamento e projeção da raiz do nariz, mandíbula proeminente, respiração bucal devido a obstrução nasal pela hiperostose; b) anormalidades neurológicas decorrentes principalmente da compressão de nervos cranianos - distúrbios visuais, surdez progressiva, paralisia facial, hemiplegia, quadriplegia, sinais de hidrocefalia ou compressão medular; c) dentição defeituosa; d) retardamento mental e motor, apenas ocasionalmente.
Os defeitos na condução auditiva podem se originar tanto da compressão do oitavo par craniano como de episódios freqüentes de otites e infecções do trato respiratório superior(11,15,16). Apesar de o paciente ter tido repetidas infecções de vias respiratórias superiores, atualmente não há indícios clínicos de déficit auditivo.
Segundo Carnevale et al.(4) e Beighton et al.(8), há grande variabilidade na expressão fenotípica da DCM. As manifestações radiográficas são mutáveis concomitantemente ao desenvolvimento corporal, sendo inicialmente mais severas na forma recessiva que na dominante. As alterações cranianas tornam-se mais pronunciadas com o tempo(10). É incerto se tais variações estão relacionadas à faixa etária ou representam graus diferentes de apresentação fenotípica(9,17).
As principais alterações radiográficas comumente descritas envolvem: hiperostose progressiva difusa dos ossos craniofaciais; espessamento e estreitamento foraminais; obliteração dos seios paranasais e células mastóideas; hipertelorismo; alargamento dos ossos curtos das mãos; esclerose inicial das diáfises com posterior alargamento gradual, afilamento cortical e "osteoporose" das metáfises dos ossos longos (aspecto em frasco de Erlenmeyer); normalidade dos ossos pélvicos, da coluna vertebral, das costelas e clavículas. Estas duas últimas localizações podem estar alargadas(11,18). Na realidade, segundo Millard e Batstone(15), os ossos são estruturalmente finos e sua radiotransparência dá a falsa impressão de osteoporose. Praticamente os achados supracitados foram encontrados na totalidade em nosso caso.
As displasias craniodiafisária e metafisária (doença de Pyle) são os principais diagnósticos diferenciais.
Os casos de DCM recessiva assemelham-se aos da displasia craniodiafisária, principalmente em relação às alterações craniofaciais, porém a avaliação radiológica dos ossos longos distingue as duas condições(19). Na DCM há alargamento metafisário e afilamento cortical determinando um padrão claviforme(19), enquanto a displasia craniodiafisária distingue-se principalmente por apresentar ossos longos retificados (cilíndricos) com aspecto de "cassetete de policial", endosteose diafisária sem expansão metafisária, convulsões e retardamento mental(2,20). Geralmente também há moderado espessamento e esclerose de costelas, clavículas e pelve(19).
Embora a DCM inicialmente apresente osteosclerose diafisária dos ossos tubulares (similar à displasia craniodiafisária), este padrão subseqüentemente desaparece, sendo substituído por expansão claviforme e afilamento cortical metafisários(18), como previamente descritos.
Em vista da considerável superposição entre a DCM recessiva e a displasia craniodiafisária, permanece em questão se ambas as condições são entidades separadas ou se são parte de um espectro da mesma doença(19). Achamos pertinente tal observação, já que, exceto pelos achados femorais, inexistência de alterações axiais, ausência de retardamento mental e de crescimento, é que pudemos enquadrar nosso caso como uma forma recessiva da DCM.
Dentre os achados radiográficos que diferenciam a doença de Pyle da DCM destacam-se mínimas ou ausentes alterações cranianas, ossos pubo-isquiáticos, costelas e clavículas marcantemente espessados, deformidade metafisária "em frasco de Erlenmeyer" mais proeminente, genu valgum e ossos da mãos com típico alargamento gradual distal dos metacarpos e proximal das falanges(21). Mediante a ausência de típicas alterações craniofaciais, além de outros achados, obrigatoriamente já se afasta a possibilidade de doença de Pyle, pois tais aspectos estão claramente presentes na DCM.
A correlação dos achados clínico-radiológicos vistos em relatos anteriores fundamenta a classificação do caso ora relatado, bem como em função de uma historia familiar aparentemente negativa e da severidade das alterações iniciais, que acreditamos tratar-se do padrão recessivo da DCM.
- 1. Jackson WP, Albright F, Dewry G, Hanelin J, Rubin MI. Metaphyseal dysplasia, epiphyseal dysplasia, diaphyseal dysplasia and related conditions. Arch Int Med 1954;94:871-85.
- 2. Richards A, Brain C, Dillon MJ, Bailey CM. Craniometaphyseal and craniodiaphyseal dysplasia, head and neck manifestations and management. J Laryngol Otol 1996;110:328-38.
- 3. Penchaszadeh VB, Gutierrez ER, Figueroa EP. Autosomal recessive craniometaphyseal dysplasia. Am J Med Genet 1980;5:43-55.
- 4. Carnevale A, Grether P, Del Castillo V, Takenaga R, Orzechowski A. Autosomal dominant craniometaphyseal dysplasia: clinical variability. Clin Genet 1983;23:17-22.
- 5. Lezzi GB, D'Elia D, Bartolini M, Camera G. Craniometaphyseal dysplasia - descrizione di un caso clinico. Pathologica 1989;81:287-90.
- 6. Haverkamp F, Emons D, Straehler-Pohl HJ, Zerres K. Craniometaphyseal dysplasia as a rare cause of a severe neonatal nasal obstruction. Int J Pediatr Otorhinolaryngol 1996;34:159-64.
- 7. Elçioglu N, Hall CM. Temporal aspects in craniometaphyseal dysplasia: autosomal recessive type. Am J Med Genet 1998;76:245-51.
- 8. Beighton P, Hamersma H, Horan F. Craniometaphyseal dysplasia - variability of expression within a large family. Clin Genet 1979;15:252-8.
- 9. Bricker SL, Langlais RP, van Dis ML. Dominant craniometaphyseal dysplasia - literature review and case report. Dentomaxillofac Radiol 1983;12:95-100.
- 10. Ramseyer LTH, Leonard JC, Stacy TM. Bone scan findings in craniometaphyseal dysplasia. Clin Nucl Med 1993;18:137-9.
- 11. Maia LC, Modesto A, Carakushansky G, Souza IPR. Craniometaphyseal dysplasia: case report. Braz Dent J 2000;11:153-60.
- 12. Taybi H, Lachman RS. Radiology of syndromes, metabolic disorders and skeletal dysplasias. 4th ed. St. Louis: Mosby, 1996.
- 13. Gorlin RJ, Spranger J, Koszalka MF. Genetic craniotubular bone dysplasia and hyperostoses - a critical analysis. Birth Defects 1969;5:79-95.
- 14. Greenspan A. Sclerosing bone dysplasias - a target-site approach. Skeletal Radiol 1991;20:561-83.
- 15. Millard RD Jr, Maisels DO, Batstone JHF, Yates BW. Craniofacial surgery in craniometaphyseal dysplasia. Am J Surg 1967;113:615-21.
- 16. Reardon W, Hall CM, Dillon MJ, Baraitser M. Sibs with mental retardation, supraorbital sclerosis, and metaphyseal dysplasia: frontometaphyseal dysplasia, craniometaphyseal dysplasia, or a new syndrome? J Med Genet 1991;28:622-6.
- 17. Spiro PC, Hamersma H, Beighton P. Radiology of the autosomal dominant form of craniometaphyseal dysplasia. S Afr Med J 1975;49:839-42.
- 18. McAlister WH, Herman TE. Osteochondrodysplasias, dysostoses, chromosomal aberrations, mucopolysaccharidoses and mucolipidoses. In: Resnick D. Diagnosis of bone and joint disorders. 3rd ed. Philadelphia: WB Saunders, 1995:4163-244.
- 19. Brueton LA, Winter RM. Craniometaphyseal dysplasia. J Med Genet 1990;27:701-6.
- 20. Schaefer B, Stein S, Oshman D, et al. Dominantly inherited craniometaphyseal dysplasia: a new craniotubular dysplasia. Clin Genet 1986;30:381-91.
- 21. Gorlin RJ, Koszalka MF, Spranger J. Pyle's disease (familial metaphyseal dysplasia). A presentation of two cases and argument for its separation from craniometaphyseal dysplasia. J Bone Joint Surg Am 1970;52:347-54.
Datas de Publicação
-
Publicação nesta coleção
02 Out 2002 -
Data do Fascículo
Jul 2002