Resumos
Leucocoria é um reflexo pupilar anormal à luz incidente, em geral relacionado a uma anormalidade intra-ocular, ocorrendo freqüentemente em crianças. A avaliação da criança com leucocoria deve ser feita principalmente para excluir o diagnóstico de retinoblastoma (causa da metade dos casos). Os autores apresentam os aspectos clínicos e de imagem essenciais ao diagnóstico das causas mais comuns de leucocoria.
Olho; Leucocoria; Infância; Ultra-sonografia; Tomografia computadorizada; Ressonância magnética
Leukokoria is an abnormal pupillary reflex most commonly seen in children that usually results from an intra-ocular abnormality. The evaluation of a child with leukokoria should be performed particularly to rule out a retinoblastoma (the cause of leukokoria in half of the cases). The authors present the main clinical and imaging features for the diagnosis of the most common causes of leukokoria.
Eye; Leukokoria; Childhood; Ultrasonography; Computed tomography; Magnetic resonance imaging
ARTIGO DE REVISÃO
Leucocoria na infância: diagnóstico diferencial por ultra-sonografia, tomografia computadorizada e ressonância magnética* * Trabalho realizado na Clínica da Imagem e no Departamento de Diagnóstico por Imagem e Anatomia Patológica do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO.
Leukokoria in childhood: differential diagnosis using ultrasonography, computed tomography, and magnetic resonance imaging
Marcelo Eustáquio Montandon JúniorI; Sizenildo da Silva FigueirêdoII; Beatriz Mahmud JacobIII; Cristiano MontandonI; Flávia Aparecida de Souza RibeiroII; Bruno Barcelos da NóbregaII; Leonardo Valadares Barbosa LôboII; Kim-Ir-Sen Santos TeixeiraIV
IMédicos Radiologistas das Clínicas Multimagem e da Imagem, Membros Titulares do Colégio Brasileiro de Radiologia e Diagnóstico por Imagem (CBR)
IIMédicos Residentes em Radiologia e Diagnóstico por Imagem do Departamento de Diagnóstico por Imagem e Anatomia Patológica da FMUFG
IIIMédica Radiologista Cooperada do Serviço de Diagnóstico por Imagem do HC-FMUFG
IVProfessor Adjunto Doutor do Departamento de Diagnóstico por Imagem e Anatomia Patológica da FMUFG
Endereço para correspondência Endereço para correspondência: Dr. Marcelo Eustáquio Montandon Júnior Rua Ismerino S. Carvalho, 775, Setor Aeroporto Goiânia, GO, 74075-040 E-mail: marmontandon@hotmail.com
RESUMO
Leucocoria é um reflexo pupilar anormal à luz incidente, em geral relacionado a uma anormalidade intra-ocular, ocorrendo freqüentemente em crianças. A avaliação da criança com leucocoria deve ser feita principalmente para excluir o diagnóstico de retinoblastoma (causa da metade dos casos). Os autores apresentam os aspectos clínicos e de imagem essenciais ao diagnóstico das causas mais comuns de leucocoria.
Unitermos: Olho; Leucocoria; Infância; Ultra-sonografia; Tomografia computadorizada; Ressonância magnética.
ABSTRACT
Leukokoria is an abnormal pupillary reflex most commonly seen in children that usually results from an intra-ocular abnormality. The evaluation of a child with leukokoria should be performed particularly to rule out a retinoblastoma (the cause of leukokoria in half of the cases). The authors present the main clinical and imaging features for the diagnosis of the most common causes of leukokoria.
Key words: Eye; Leukokoria; Childhood; Ultrasonography; Computed tomography; Magnetic resonance imaging.
Introdução
Leucocoria (das palavras gregas leukos = branco e koria = pupila) corresponde a um reflexo pupilar anormal branco, róseo ou amarelo esbranquiçado(1). Este sinal clínico sugere a presença de anormalidade anterior à retina, que reflete a luz incidente à pupila antes que a luz alcance a retina ou a coróide, podendo decorrer de tumor intra-ocular, membrana vítrea ou descolamento retiniano(1).
A causa mais comum de leucocoria é o retinoblastoma, um tumor de células retinianas imaturas. Outras causas comuns, em ordem decrescente de freqüência, são a persistência hiperplásica do vítreo primitivo (PHVP), a doença de Coats, a toxocaríase, a retinopatia da prematuridade (RP) e o hamartoma astrocítico(1). Causas menos freqüentes são os colobomas de coróide, as hemorragias vítreas organizadas e o meduloepitelioma. Catarata congênita e descolamento da retina de longa evolução também devem ser lembrados. Não foi objetivo do estudo enfocar as causas menos freqüentes de leucocoria.
Além de uma adequada análise semiológica, incluindo exame oftalmoscópico, a utilização de métodos de imagem torna-se imperativa para a realização de um diagnóstico preciso.
A ultra-sonografia (US) torna-se extremamente útil quando a avaliação oftalmoscópica é difícil ou impossível diante de um meio ocular opaco impedindo a condução da luz, bem como na caracterização de tumores intra-oculares suspeitados.
Em função da alta sensibilidade para detecção de calcificação, a tomografia computadorizada (TC) é exame importante para o diagnóstico e estadiamento dos retinoblastomas (tumores freqüentemente calcificados).
A ressonância magnética (RM) pode ser usada como método complementar à TC se há tumor ocular, coleções líquidas anormais ou descolamento retiniano(1). O uso do gadolínio revela áreas de impregnação anormal e seqüências gradiente-eco também podem identificar calcificações.
Neste artigo revisamos as principais causas de leucocoria na infância, enfatizando alguns aspectos clínicos e achados de imagem essenciais a uma adequada elucidação diagnóstica (Quadro 1).
RETINOBLASTOMA (Quadro 2)
Retinoblastoma é o tumor intra-ocular primário maligno mais comum em crianças e corresponde a 30% de todos os tumores oculares. Ocorre em cerca de 1:20.000 nascidos e é responsável por 1% de todos os tumores infantis e metade dos casos de crianças com leucocoria(1).
Origina-se de células imaturas derivadas do epitélio embriogênico retiniano da vesícula óptica primária, destinadas a se tornarem fotorreceptores da retina(2).
A etiologia não é conhecida, embora se manifeste de forma hereditária (autossômica dominante), não-hereditária e por deleção cromossômica(2). Ambos os sexos são igualmente afetados(1,2) e 25% a 30% dos casos têm doença bilateral(3).
No início os tumores são vistos à fundoscopia e mais tarde a leucocoria e olho de "gato" amaurótico são evidentes(2). A maioria (cerca de 80%) tem o diagnóstico antes dos 3-4 anos de vida(3), sendo a média 18 meses(2). Nos países em desenvolvimento, como o Brasil, o diagnóstico é tardio, fazendo com que quase 40% dos casos sejam extra-oculares ao diagnóstico(3).
A variedade endofítica do tumor, que tem crescimento centrípeto, projeta-se da retina para dentro da cavidade vítrea(2), podendo também atingir a câmara anterior(1), com aspecto de área branca ou rosada demarcada pela presença de calcificações secundárias(2). A forma exofítica cresce no espaço sub-retiniano, causando elevação e descolamento da retina, e neste caso a US pode revelar um tumor oculto nesta localização(1,2).
A US demonstra estar o tumor situado em uma única área do globo ocular ou ser mais extenso. Depósitos de cálcio podem estar presentes como focos hiper-refringentes e com sombra acústica posterior, auxiliando no diagnóstico(2). O contorno tumoral é freqüentemente irregular e algumas lesões císticas reconhecidas como coágulos de sangue ou necrose podem ocorrer. É importante a detecção de invasão ao longo do nervo óptico para que este seja excisado durante a enucleação(2).
A avaliação da extensão tumoral às porções laterais e mediais da órbita e às regiões extra-oculares é bastante limitada no exame pelo ultra-som(4).
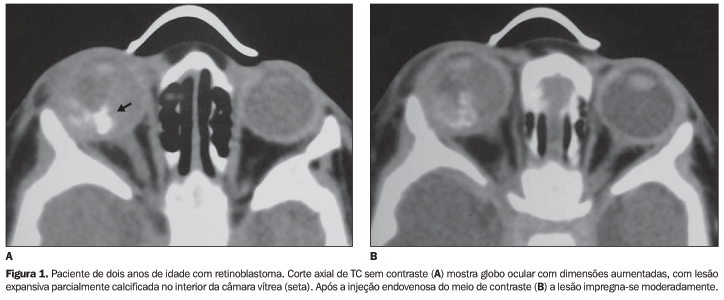
Uma área intra-ocular hiperdensa contendo calcificações nodulares ou puntiformes em mais de 90% dos casos é o aspecto visto à TC(1,2,4,5). A presença, tamanho e aspecto das calcificações dependem da dimensão do tumor(6) (Figuras 1 e 2).
Sabe-se que um retinoblastoma de pequenas dimensões é desprovido de calcificação e que em crianças menores de três anos a evidenciação tomográfica de calcificação tumoral é virtualmente diagnóstica(5,6). Esta geralmente localiza-se na porção posterior do globo, com extensão para o vítreo(5). Pacientes com leucocoria sem calcificação evidente à TC provavelmente têm lesão que simula o retinoblastoma(6).
Este método diagnóstico também evidencia mais claramente o envolvimento extra-ocular e do nervo óptico, além de ser essencial para mostrar a disseminação pelo sistema nervoso central ou o envolvimento da glândula pineal por um pinealoma(2,7), caracterizando o chamado "retinoblastoma trilateral"(7). Além de mesma origem neuroectodérmica, a glândula pineal tem função fotorreceptora em vertebrados inferiores e a histologia do pinealoma pode não ser distinguida de um retinoblastoma(4).
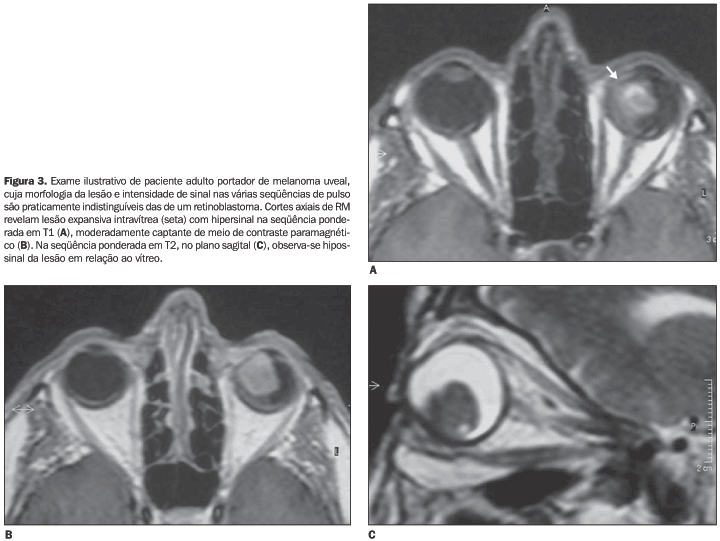
Na RM os retinoblastomas possuem iso a hipersinal, em relação ao vítreo, nas seqüências ponderadas em T1 e DP, e hipossinal em T2(1). As calcificações podem ser vistas como variáveis graus de hipointensidade em todas as seqüências de pulso (melhor identificadas nas seqüências gradiente-eco) e estes tumores mostram moderado a intenso realce em imagens pós-contraste paramagnético(4)(Figura 3). O exsudato sub-retiniano secundário possui hipersinal em T1 e T2, devido ao seu conteúdo proteináceo(1).
Metástases de retinoblastoma aparecem dentro de dois anos após o diagnóstico(1). Quando o tumor se estende além do olho, a mortalidade aproxima-se de 100%. A invasão dos vasos da coróide aumenta em até 24% a taxa de mortalidade(1).
Comparativamente à TC, a RM fornece mais informações para a diferenciação do retinoblastoma com outras afecções intra-oculares causadoras de leucocoria(4).
PERSISTÊNCIA HIPERPLÁSICA DO VÍTREO PRIMITIVO (Quadro 3)
A PHVP ocorre quando há falha na regressão normal do sistema vascular hialóide embrionário (canal de Cloquet), sendo a lesão básica causada pela persistência de várias porções do vítreo primitivo e da túnica vasculosa lentis, que sofrem hiperplasia devido a uma intensa proliferação fibrovascular de tecido conectivo embriogênico(4,8).
Não há predileção por sexo e é geralmente detectada em recém-natos prematuros(9). É unilateral em 90% dos casos, cursando com leucocoria, microftalmia e, por vezes, estrabismo e nistagmo(10).
A etiologia permanece desconhecida na maioria dos casos, porém a presença de alguns raros relatos em familiares sugere a possibilidade de herança autossômica dominante ou recessiva(9). A PHVP pode ser uma das manifestações de doença mais extensa, como a doença de Norrie (degeneração óculo-acústico-cerebral progressiva), a síndrome de Warburg (más-formações congênitas do sistema nervoso central, microftalmia e descolamento de retina uni ou bilateral), a displasia vítreo-retiniana primária ou outros defeitos congênitos(4).
A PHVP pode mostrar três padrões distintos: uma variante anterior, outra posterior e uma terceira, denominada completa (somatório das duas primeiras)(7).
A forma mais típica é a anterior (25% dos casos), em que uma placa de tecido conectivo vascularizado está situada no vítreo, imediatamente atrás e aderente ao cristalino, conectando-se perifericamente e tracionando os processos ciliares anormalmente alongados(7). A porção anterior da placa pode distender a cápsula do cristalino e distorcer sua morfologia(7).
A forma puramente posterior da PHVP (12% dos casos) ocorre em globos com anormalidades confinadas ao segmento ocular posterior, tais como pregas retinianas, traves e membranas vítreas, anormalidades maculares e do nervo óptico(9). Nesta variante, lesão retrolental, a artéria hialóide e uma trave de tecido conectivo são freqüentemente encontradas na câmara vítrea, estendendo-se em todo o trajeto do disco óptico(9). Opostamente à forma anterior, as estruturas do pólo ventral do globo estão poupadas, tais como a câmara anterior e o cristalino(9).
A combinação das apresentações anterior e posterior é a variante clínica mais freqüentemente encontrada(9) , correspondendo a aproximadamente dois terços dos casos de PHVP.
O estiramento dos processos ciliares é achado quase patognomônico, e sinéquias irido-cristalinianas podem ser observadas em alguns casos(10). Más-formações retinianas e descolamento da retina podem estar presentes(8), sendo este último gerado pela tração da trave fibrovascular no pólo posterior do globo(9), porém, extensos descolamentos da retina são raros.
À US o globo ocular afetado pode parecer menor do que o normal(11). O aspecto típico é um triângulo hiperecogênico cuja base se situa na face posterior do cristalino e o ápice, no nível da papila óptica (Figura 4). Outros achados incluem a presença de tumoração ecogênica retrolental ou múltiplos ecos intravítreos(10), causados por hemorragia. Câmara posterior ecograficamente normal pode ser observada, porém é raro(10). O uso do método Doppler pode revelar fluxo sanguíneo na artéria hialóide persistente. Este achado ao Doppler, associado a hemorragia vítrea e a ausência de calcificação, é virtualmente patognomônico de PHVP(12).
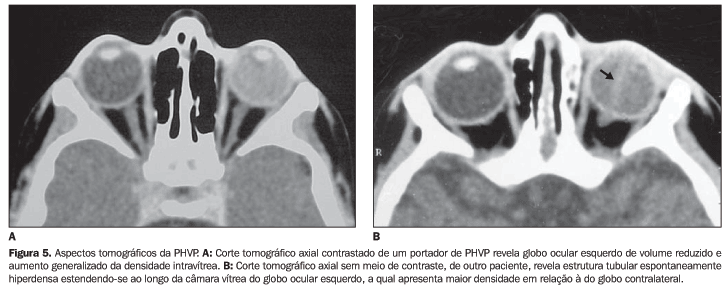
Os achados tomográficos comumente observados na PHVP incluem(4,8): microftalmia; ausência de calcificações; aumento difuso da densidade da câmara vítrea; imagem tubular, triangular retrolental ou outra densidade intravítrea inespecífica; realce anormal do tecido intravítreo na fase pós-contraste endovenoso; lente pequena ou irregular e câmara anterior rasa.
À mudança de decúbito pode-se observar deslocamento gravitacional do nível líquido dentro da câmara vítrea, refletindo fluido sero-sanguinolento no espaço sub-hialóide ou no espaço sub-retiniano (Figura 5).
A RM pode demonstrar os vários aspectos inerentes à PHVP, sendo, portanto, altamente específica para o diagnóstico desta entidade(7). A especificidade diagnóstica depende da demonstração do baixo sinal da placa fibrovascular retrolental, em íntima associação com anormalidades na configuração do cristalino, dentro de um pequeno globo ocular(7). O descolamento da retina pode ser evidenciado.
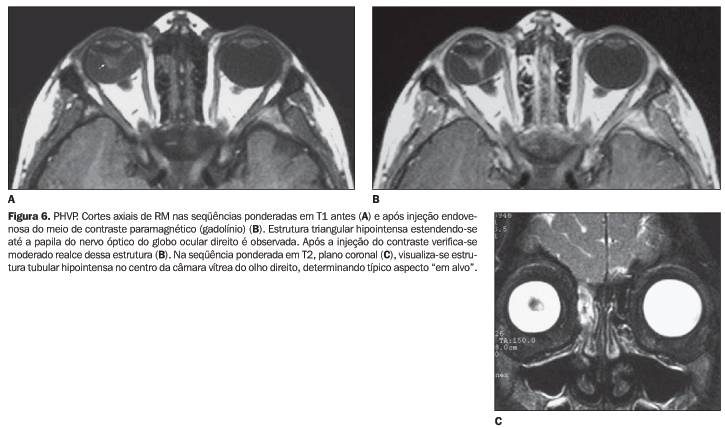
A apresentação clássica do remanescente vascular hialóide nos exames de RM inclui uma estrutura linear hipointensa nas seqüências ponderadas em T1 estendendo-se diretamente da parte posterior do cristalino até a papila do nervo óptico(7). A câmara vítrea costuma ser hiperintensa nas imagens ponderadas em T1 e em T2(7,8), devido à hemorragia(7). A placa retrolental, geralmente bem vascularizada, pode apresentar realce pelo meio de contraste paramagnético (gadolínio)(7) (Figura 6).
A configuração do cristalino comumente é anormal na PHVP. Porém, se esta for normal, na presença de lesão retrolental, um outro diagnóstico deve ser fortemente considerado, como o retinoblastoma(7).
A aparência da RM na retinopatia do prematuro pode ser similar à da PHVP(4), pois tais alterações podem coexistir.
DOENÇA DE COATS (Quadro 4)
É anomalia vascular primária idiopática da retina, correspondendo a 16% dos casos de leucocoria(1), e caracterizada por telangiectasias e descolamento retiniano exsudativo secundário(4,13). Freqüentemente estes vasos anômalos mostram áreas de dilatação aneurismática(1).
Sua etiologia permanece incerta(14) e estudos com microscopia óptica e eletrônica têm mostrado a perda da barreira endotelial vascular (hemato-retiniana) como o defeito básico da doença, levando a anormalidades secundárias, como exsudato intra-retiniano, depósitos lipídicos, acúmulo de macrófagos repletos de lipídios (células fantasmas), cristais de colesterol e hemorragia(14). O exsudato tem alto teor protéico e raramente pode ser rico em lipídios(1).
Embora presente ao nascimento, a anomalia vascular da doença de Coats comumente não causa sintomas até que ocorra descolamento retiniano e perda da visão central(4,13). Este descolamento é freqüentemente total e completo em casos avançados da doença(1). A retina temporal é amiúde mais afetada que a nasal(14).
A doença de Coats é classificada em cinco estágios: estágio 1 - apenas telangiectasias; estágio 2 - telangiectasias e exsudação; estágio 3 - descolamento da retina; estágio 4 - descolamento total e glaucoma secundário; estágio 5 - doença em fase avançada irreversível. Os estágios 4 e 5 são de péssimo prognóstico(15).
Clinicamente, os pacientes podem apresentar estrabismo, leucocoria, perda da visão (anopsia) e glaucoma secundário(1). As demonstrações de exsudato sub-retiniano amarelado e telangiectasia periférica são virtualmente patognomônicas para este diagnóstico(14).
O pico etário para o surgimento dos sinais e sintomas situa-se entre os seis e oito anos(1,15-17). A doença é quase sempre unilateral e afeta duas vezes mais meninos que meninas(1).
A US pode demonstrar o descolamento da retina(1,14) e o material sub-retiniano pode conter ecos particulados suspensos, presumivelmente correspondentes a cristais de colesterol(1,17).
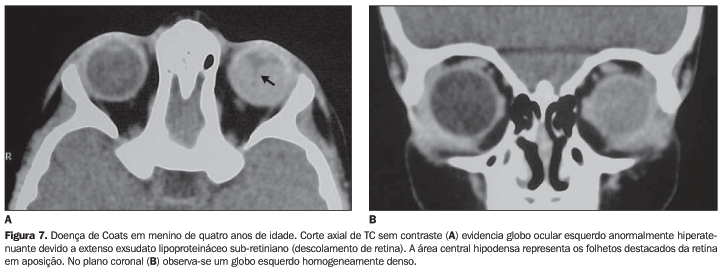
A TC costuma revelar aumento difuso da atenuação no globo, em virtude da densidade do exsudato proteináceo sub-retiniano, e calcificações são extremamente raras(1) (Figura 7).
A RM é superior à TC na diferenciação da doença de Coats em relação ao retinoblastoma e outras causas de leucocoria(1). Este método também demonstra o descolamento da retina, podendo este apresentar hipersinal nas ponderações em T1 e T2, devido à mistura lipoprotéica do exsudato(1,17) (Figura 8).
Ocasionalmente, o exsudato pode mostrar hipointensidade em T2 relativamente ao vítreo, em função da significante quantidade de cristais de colesterol e lipídios de membrana(1,17). A retina descolada pode revelar realce após injeção do contraste paramagnético (gadolínio)(1,4,17), todavia, não há realce no exsudato sub-retiniano e nenhuma área captante está presente(4).
TOXOCARÍASE OCULAR (Quadro 5)
A toxocaríase ocular ou endoftalmite por Toxocara é tipicamente uma doença monocular de crianças e seus achados clínicos incluem coriorretinite posterior e periférica, papilite óptica e endoftalmite. A inflamação resultante do envolvimento ocular pode resultar em formação de uma membrana sobre a retina, tração e descolamento retinianos(18).
Toxocara canis e Toxocara catis são helmintos nematódeos(1), agentes infecciosos da toxocaríase(19). Os cães e os gatos, hospedeiros definitivos do parasita, contaminam os solos ao eliminarem ovos de Toxocara por suas fezes(2,18,19).
A forma de transmissão pode ser feita de maneira direita ou indiretamente(19), através da ingestão de ovos provenientes de solos contaminados(5) ou ingestão de leguminosas cruas e mal lavadas(19). As crianças, em idade pré-escolar e que apresentam comportamento geófago, entram em contato com as larvas(18,19). O comprometimento da larva migrans ocular é mais freqüente nas crianças em idade escolar(19). Lesões oculares aparecem em meses a anos após a infecção inicial(5).
As infecções por T. canis são muito mais freqüentes(19) e responsáveis por 16% das causas de leucocoria em crianças(1,20).
A infecção apresenta-se sobre duas formas distintas: larva migrans visceral ou larva migrans com o comprometimento ocular; as duas formas decorrem da migração da larva pelo organismo(19). Do intestino a larva chega ao fígado, e do fígado pode migrar para os pulmões e outros tecidos, incluindo o cérebro e olhos(1). A forma visceral provoca hipereosinofilia, hepatoesplenomegalia, pneumonite, febre e hiperglobulinemia, e os sintomas variam de acordo com o órgão acometido(19). O acometimento ocular (endoftalmite) acarreta leucocoria e baixa da acuidade visual do olho afetado(1,18), dor ocular e estrabismo(19), inflamação e opacificação vítrea(5), bem como eosinofilia periférica(1).
A reação inflamatória usualmente é acompanhada pelo acúmulo de "débris" dentro do vítreo (vitreíte)(1) e o material vítreo-retiniano pode "encolher" criando tração e produzindo descolamento secundário da retina e catarata. O processo de tração é incomum no retinoblastoma, mas pode ocorrer na PHVP(19).
Os achados patológicos revelam que, microscopicamente, a retina está elevada e distorcida, como também pode estar deslocada por um conglomerado de processo inflamatório que contém tecido cicatricial abundante e denso. A larva do T. canis está usualmente envolta dentro dessa massa retiniana, evitando sua visualização direta(1). A cicatriz pode exercer tração na retina e haver um exsudato sub-retiniano proteináceo oriundo da reação inflamatória que a larva causa(1). A coróide subjacente está infiltrada por células inflamatórias com eosinófilos.
O diagnóstico laboratorial de infecção por Toxocara é feito pelo método Elisa, com sangue periférico ou aspirado vítreo, que revela presença de eosinófilos na fase ativa da inflamação (não específicos da toxocaríase)(1). O diagnóstico específico do T. canis como causa de endoftalmite requer identificação histológica do organismo(1). Por se tratar de uma afecção com processo basicamente inflamatório, o tratamento geralmente é feito com a administração de corticóides.
Os achados de imagem ajudam, principalmente, excluir o retinoblastoma(20) e outras afecções como causa de leucocoria(1).
Na US observa-se lesão periférica intra-ocular hiperecogênica(1,20). Descolamento de retina e ecos internos em suspensão dentro do vítreo podem ser identificados, mas não são achados específicos(1,20). Calcificação é incomum(1).
A TC mostra olho de tamanho normal e sem calcificações, revelando ainda lesão intravítrea com espessamento e inflamação da esclera(1,5,20). A reação granulomatosa ao redor da larva aparece como espessamento focal úveo-escleral(1,5). Tração e descolamento secundários da retina podem ser evidentes pela TC(1,5), e aumento difuso da densidade dentro do globo ocular sem calcificações pode ser visto(5).
As imagens obtidas por RM comumente revelam lesão central ou periférica com intensidade de sinal variável(20). Nas seqüências ponderadas em T1 o sinal é isointenso em relação ao vítreo e em T2 é hiperintenso(20), diferentemente do retinoblastoma. Contudo, em alguns casos a "massa inflamatória" pode ser hipointensa em T2 devido à presença de tecido fibroconectivo denso que se forma com o granuloma(1). Os diferentes achados de intensidade de sinal são decorrentes do conteúdo protéico e/ou hemorragia associada(5).
O exsudato sub-retiniano pode apresentar-se hiperintenso nas seqüências ponderadas em T1 e em T2. Após injeção de contraste paramagnético, a RM pode demonstrar múltiplas áreas de moderado a intenso realce anômalo(1) na câmara vítrea e nas paredes do globo ocular afetado, usualmente espessadas(20).
RETINOPATIA DA PREMATURIDADE (Quadro 6)
A RP corresponde a uma organização fibrovascular pós-natal do humor vítreo, comumente resultando no descolamento da retina(1,17). Esta condição representa 3% a 5% dos casos de leucocoria na infância(1) e os fatores essenciais desencadeantes da RP parecem ser a prematuridade e o uso de oxigenoterapia. Quanto menor a criança, maior o risco de desenvolver a doença(4).
Três estágios de RP são evidenciados à avaliação oftalmoscópica. No estágio ativo ou precoce há estreitamento arteriolar causado por espasmo dos vasos retinianos em resposta à hiperoxigenação. No segundo, depois de retirada a oxigenoterapia, os vasos tornam-se tortuosos, dilatados e alongados. No terceiro estágio alguns desses novos vasos gradualmente crescem da retina para o humor vítreo, causando hemorragia vítrea e descolamento retiniano(1,17).
Quanto ao prognóstico, na maioria dos pacientes (85% a 95%) a neovascularização vítrea regride espontaneamente(1,4), todavia, em alguns pacientes a retinopatia progride para estágio cicatricial caracterizado pela formação de densa membrana de tecido vascularizado cinza-esbranquiçado retrolental e por retina deslocada com formação de cicatrizes. Tal processo inibe o crescimento normal dos globos oculares, produzindo microftalmia bilateral, geralmente assimétrica(1,17).
A presença de microftalmia depende da idade do paciente à avaliação, da gravidade e do estágio da doença(1). Ocasionalmente a RP pode apresentar-se como leucocoria unilateral(4) e um sistema vascular hialóide persistente pode ser um achado associado em pacientes com RP(1,21). Em sete de 14 pacientes portadores de RP estudados por Eller et al.(21), um sistema vascular hialóide persistente foi observado.
A US poderá evidenciar microftalmia bilateral, descolamento retiniano e lesão intra-ocular periférica no pólo temporal (área onde a vascularização desenvolve-se por último)(1,17).
Na TC o globo inteiro freqüentemente acha-se hiperatenuante devido à neovascularização interna(1). Calcificação é rara na RP(4), porém calcificações distróficas podem eventualmente ocorrer em estágios tardios da doença(1,4). Microftalmia e câmara anterior rasa também podem ser vistas nos estágios avançados(1,4,17) (Figura 9).
Nas imagens ponderadas em T1 e T2, globos microftálmicos na RP podem demonstrar hiperintensidade, devido às hemorragias sub-retinianas crônicas(1,17). Concomitantemente, "massa" retrolental pode ser observada relativamente hipointensa ao líquido sub-retiniano hiperintenso. Tal aspecto representa a aposição dos folhetos destacados da retina(1,17).
História de prematuridade, tratamento em incubadoras, bilateralidade e achados oftalmoscópicos, ecográficos e tomográficos são geralmente suficientes para estabelecer o diagnóstico de RP.
HAMARTOMAS ASTROCÍTICOS (Quadro 7)
Hamartomas astrocíticos ou astrocitomas retinianos são neoplasias incomuns originadas da camada de fibras nervosas da retina ou do nervo óptico(1,4), embora os hamartomas dos nervos ópticos raramente ocorram(22). Representam quase 3% dos casos de leucocoria na infância(1,17).
Esses hamartomas retinianos tipicamente não prejudicam a visão, a menos que acometam a mácula e causem hemorragia vítrea(23). Opostamente a outras lesões retinianas, o hamartoma é incomumente complicado por exsudato sub-retiniano (que pode provocar descolamento retiniano)(1), bem como hemorragia vítrea(24).
Podem ser encontrados como lesão solitária incidental em indivíduos normais, raramente em pacientes com neurofibromatose, porém estão mais freqüentemente associados a esclerose tuberosa(24).
Mais de 50% dos pacientes com esclerose tuberosa têm um ou mais desses tumores retinianos(17,22). Em metade desses pacientes os hamartomas ocorrem bilateralmente(22) e a concomitância de dois ou mais tipos morfológicos tem sido observada(25). Cerca de 14% ocorrem em pacientes com neurofibromatose tipo 1 (doença de von Recklinghausen) e são esporádicos em um terço dos casos(17).
Três tipos morfológicos básicos de hamartomas são reconhecidos. O tipo mais comum consiste em lesão oval ou circular, semitransparente e com tonalidade salmão, relativamente plana e com superfície lisa, localizada na retina, mais comumente próxima ou no pólo posterior. O segundo tipo mais freqüente é lesão multinodular calcificada, elevada, branca e opaca, semelhante a uma amora. O terceiro contém características de ambas, sendo um hamartoma plano com área central nodular e calcificada, enquanto sua periferia é semitransparente, lisa e com tonalidade salmão(22,25).
As lesões geralmente não crescem, porém tardiamente algumas podem tornar-se calcificadas(22). Casos de regressão da lesão têm sido observados(24) e não há correlação entre a idade do paciente e a presença de hamartomas astrocíticos(25).
Presumivelmente, as lesões calcificadas facilmente serão identificadas à US em aposição à retina posterior como foco fortemente ecogênico, comumente associado a sombra acústica posterior.
Na TC, uma única ou múltiplas lesões podem ser identificadas. Variavelmente, podem mostrar realce após administração do meio de contraste e calcificações são ocasionalmente notadas. Adicionalmente, os cortes tomográficos do cérebro podem mostrar características típicas da esclerose tuberosa, bem como da neurofibromatose tipo 1(1)(Figura 10).
Como o aspecto usual é de nódulo único ou de múltiplos nódulos elevados 1 mm ou 2 mm acima da superfície da retina, a TC e a RM podem não evidenciar as lesões nesse estágio(1). Porém, quando acima de 3 mm, costumam ser vistos por esses métodos. Ocasionalmente, o aspecto tomográfico dos hamartomas astrocíticos pode simular o retinoblastoma(17).
DIAGNÓSTICO DIFERENCIAL
O objetivo primordial é excluir ou confirmar casos de retinoblastoma, em função das importantes repercussões causadas por esta neoplasia, necessitando de terapia específica (ressecção). A seqüência de investigação pelos métodos de imagem deve basear-se na adequada coleta de informações clínicas e na avaliação oftalmoscópica, objetivando maximizar a sensibilidade desses métodos em relação aos aspectos macroscópicos esperados, mediante a hipótese clínica aventada.
A combinação de dados clínicos e achados de imagem torna freqüentemente possível o diagnóstico específico em casos de leucocoria na infância (Tabela 1).
Recebido para publicação em 18/3/2003. Aceito, após revisão, em 3/9/2003.
- 1.Smirniotopoulos JG, Bargallo N, Maffee MF. Differential diagnosis of leukokoria: radiologic-pathologic correlation. RadioGraphics 1994;14:1059-79.
- 2.Fielding JA. The eye and orbit. In: Cosgrove D, Meire H, eds. Abdominal and general ultrasound. Edinburgh: Churchill Livingstone, 1993:621-58.
- 3.Centro de Tratamento e Pesquisa Hospital do Câncer A.C. Camargo. Retinoblastoma, 2001. (http://www.hcanc.org.br)
- 4.Maffe MF. Eye and orbit. In: Som PM, Curtin HD, eds. Head and neck imaging. 3rd ed. St. Louis: Mosby, 1996:1109-37.
- 5.Grossman RI, Yousem DM. Orbit. In: Grossman RI, Yousem DM, eds. The requisites - neuroradiology. St. Louis: Mosby, 1994:284-6.
- 6.Char DH, Hedges TR III, Norman D. Retinoblastoma. CT diagnosis. Ophthalmology 1984;91:1347-50.
- 7.Atlas SW, Galetta SL. The orbit and visual system. In: Atlas SW, ed. Magnetic resonance imaging of the brain and spine. 2nd ed. Philadelphia: Lippincott Williams & Wilkins, 1996:1026.
- 8.Mafee MF, Goldberg MF. Persistent hyperplastic primary vitreous (PHPV): role of computed tomography and magnetic resonance. Radiol Clin North Am 1987;25:683-92.
- 9.Silbert M, Gurwood AS. Persistent hyperplastic primary vitreous. Clin Eye Vis Care 2000;12:131-7.
- 10.Monnot JP, Assi A. Persistence et hyperplasie du vitré primitif: resultats à moyen terme de la vitrectomie. J Fr Ophtalmol 1992;15:269-73.
- 11.Sallum JMF, Moreira Jr CA. Anomalias vítreas congênitas. In: Abujamra S, Ávila M, Barsante C, et al Retina e vítreo - clínica e cirurgia. São Paulo: Roca, 2000:245-61.
- 12.Mihmanli I, Albayram MS, Kantarci F, Adaletli I, Islak C, Altug A. Persistent hyperplastic primary vitreous and von Hippel-Lindau disease: contribution of color Doppler ultrasonography. J Ultrasound Med 2002;21:565-8.
- 13.Sherman JL, McLean IW, Brallier DR. Coats' disease: CT-pathologic correlation in two cases. Radiology 1983;146:77-8.
- 14.Char DH. Coats' syndrome: long term follow-up. Br J Ophthalmol 2000;84:37-9.
- 15.Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats disease: the 2000 proctor lecture. Am J Ophthalmol 2001;131:572-83.
- 16.Burgener FA, Kormano M. Diagnóstico diferencial em tomografia computadorizada. Rio de Janeiro: Revinter, 1998.
- 17.Dähnert W. Radiologia - manual de revisão. Rio de Janeiro: Revinter, 2001.
- 18.Sabrosa NA, Souza EC. Nematode infections of the eye: toxocariasis and diffuse unilateral subacute neuroretinitis. Curr Opin Ophthalmol 2001;12:450-4.
-
19Bureau de la sécurité de laboratoires. Fiche technique santé-sécurité-matières infectieuses: Toxocara canis/Toxocara catis, 2001. (http://hc-sc.gc.ca.org)
- 20.Abbehusen C, Jesus PEM, Rodrigues WM, Wolosker AMB, Lederman HM, Yamashita HK. Toxocaríase ocular: relato de caso por ressonância magnética e revisão da literatura. Rev Imagem 1998;20:163-6.
- 21.Eller AW, Jabbour NM, Hirose T, Schepens CL. Retinopathy of prematurity: the association of a persistent hyaloid artery. Ophthalmology 1987;94:444-8.
- 22.Robertson DM. Ophthalmic manifestations of tuberous sclerosis. Ann N Y Acad Sci 1991;615:17-25.
- 23.Dotan SA, Trobe JD, Gebarski SS. Visual loss in tuberous sclerosis. Neurology 1991;41:1915-7.
- 24.Kirath H, Belgiç S. Spontaneous regression of retinal astrocytic hamartoma in a patient with tuberous sclerosis. Am J Ophthalmol 2002;133:715-6.
- 25.Rowley SA, O'Callaghan FJ, Osborne JP. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol 2001;85:420-3.
Datas de Publicação
-
Publicação nesta coleção
31 Maio 2004 -
Data do Fascículo
Abr 2004
Histórico
-
Recebido
18 Mar 2003 -
Aceito
03 Set 2003