Abstract
The clinical management and decision-making in pregnancies in which there is suspicion of lethal fetal malformations during the prenatal period, such as lethal skeletal dysplasia (SD), demand a multidisciplinary approach coordinated by an experienced physician. Based on the presentation of a case of osteogenesis imperfecta type IIA, we offer and discuss recommendations with the intention of organizing clinical and laboratory investigations aiming toward the clinical management, prognosis, and etiological diagnosis of these malformations, as well as genetic counselling to patients who wish to become pregnant.
Keywords:
pregnancy; skeletal dysplasia; osteogenesis imperfecta; prenatal period; genetic syndromes
Resumo
O manejo clínico e a tomada de decisões médicas em gestantes com suspeita de malformação letal em um feto no período pré-natal, tal qual uma displasia esquelética letal, demandam uma abordagem multidisciplinar coordenada por um médico experiente. Baseado na apresentação de um caso de osteogênese imperfeita tipo IIA, recomendações são apresentadas e discutidas com a intenção de organizar as investigações clínicas e laboratoriais visando o manejo clínico, o prognóstico, e o diagnóstico etiológico dessas malformações, e o aconselhamento genético para as pacientes que desejam engravidar.
Palavras-chave:
gravidez; displasia esquelética; osteogênese imperfeita; período pré-natal; síndromes genéticas
Introduction
Skeletal dysplasias (SDs), or osteochondrodysplasias (OCDs), are a group of bone disorders with clinical and etiological heterogeneous characteristics. They affect the bone tissue and cartilage, resulting in changes in the growth, shape and development of the skeletal system. The OCDs can be very rare; however, as a group, their prevalence is estimated at around 2.4 per 10,000 live births11. Camera G, Mastroiacovo P. Birth prevalence of skeletal dysplasias in the Italian Multicentric Monitoring System for Birth Defects. Prog Clin Biol Res 1982;104:441-449 22. Rasmussen SA, Bieber FR, Benacerraf BR, Lachman RS, Rimoin DL, Holmes LB. Epidemiology of osteochondrodysplasias: changing trends due to advances in prenatal diagnosis. Am J Med Genet 1996;61(01):49-58, with the lethal SD forms corresponding to 0.95-1.5 per 10,000 births.11. Camera G, Mastroiacovo P. Birth prevalence of skeletal dysplasias in the Italian Multicentric Monitoring System for Birth Defects. Prog Clin Biol Res 1982;104:441-449 22. Rasmussen SA, Bieber FR, Benacerraf BR, Lachman RS, Rimoin DL, Holmes LB. Epidemiology of osteochondrodysplasias: changing trends due to advances in prenatal diagnosis. Am J Med Genet 1996;61(01):49-58 33. Andersen PE Jr. Prevalence of lethal osteochondrodysplasias in Denmark. Am J Med Genet 1989;32(04):484-489 It is known that there are more than 456 entities classified into 40 categories by their cardinal features (radiological findings, molecular etiology, inheritance), among which 40% can be already detected in the perinatal period, representing 9 deaths per 1,000 births.11. Camera G, Mastroiacovo P. Birth prevalence of skeletal dysplasias in the Italian Multicentric Monitoring System for Birth Defects. Prog Clin Biol Res 1982;104:441-449 44. Orioli IM, Castilla EE, Barbosa-Neto JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet 1986;23(04):328-332
Osteogenesis imperfecta type II (OI type II) comprises 14% of lethal SDs, and is the second most common cause, among thanatophoric dysplasia (26%) and the achondrogenesis (9%) group, which represents 40–60% of all lethal SDs.11. Camera G, Mastroiacovo P. Birth prevalence of skeletal dysplasias in the Italian Multicentric Monitoring System for Birth Defects. Prog Clin Biol Res 1982;104:441-449 55. Krakow D, Alanay Y, Rimoin LP, et al. Evaluation of prenatal-onset osteochondrodysplasias by ultrasonography: a retrospective and prospective analysis. AmJMed Genet A2008;146A(15):1917-1924 66. Tretter AE, Saunders RC, Meyers CM, et al. Antenatal diagnosis of lethal skeletal dysplasias. Am J Med Genet 1998;75(05):518-522 77. Källén B, Knudsen LB, Mutchinick O, et al. Monitoring dominant germ cell mutations using skeletal dysplasias registered in malformation registries: an international feasibility study. Int J Epidemiol 1993;22(01):107-115 Osteogenesis imperfecta type II is a genetic disorder of the connective tissue characterized by severe bone fragility, susceptibility to severe deformities, and the occurrence of several pathological fractures, with predominance of de novo autosomal dominant inheritance caused by mutations in genes COL1A1 and COL1A2.88. Basel D, Steiner RD. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet Med 2009;11(06):375-385 Multiple fractures are frequent in utero, and perinatal death occurs in the majority of cases,99. Huber MA. Osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;103(03):314-320 where most prenatal diagnosis are suspected primarily by early fetal ultrasound, and confirmed by skeletal radiography, autopsy and, less frequently, by molecular tests.
Due to the suspicion of a presumptive diagnosis of SD based on early gestational ultrasound findings, a range of clinical diagnosis with different outcomes may be considered. Based on the severity of the condition, such considerations and observations can impose difficulties to an appropriate clinical management of these fetuses, the pregnant women, and their families regarding genetic counselling.
In face of a medical emergency for the fetus with a presumed lethal SD, and the presence of a rather uniform gestational ultrasound phenotype among lethal SDs, clinical management guidelines become crucial. They may assist physicians to define proper etiological diagnosis and clinical prognosis, as well as genetic counselling, once few of these genetic disorders may present with considerable recurrence risks.
This article aims, through the presentation of a clinical case of a lethal SD with radiological features (OI type IIA), to review the most common lethal SDs in the perinatal period, highlight their clinical and radiologic features, and compare the reported case with the literature. Furthermore, recommendations for the clinical management of similar cases are discussed.
Clinical Case Description
A 33-year-old pregnant woman in her 6th pregnancy, with 5 previous vaginal deliveries, was referred to our outpatient prenatal clinic due to a malformation in her fetus detected by an ultrasound in the Family Care Clinic. Her family history was unremarkable. A prenatal ultrasound fetal biometry at 23 weeks of pregnancy revealed an estimated fetal weight of 505 g and shortening of the long bones, so the hypothesis of an SD was considered. At 39 weeks of pregnancy, an ultrasound revealed an estimated fetal weight of 815 g and fetal malformations characterized by hypotelorism, short and saddled nose, micrognathia with redundancy of soft tissue in the face and neck, very short ribs, and narrow thoracic cage (Fig. 1a). Furthermore, it was possible to observe skeletal abnormalities with shortening of the limbs more pronounced in the femora and humeri. Fetal hypocalcification of the skull was evident, and complete and normal visualization of the encephalon, cerebral hemispheres, ventricles and the posterior fossa was apparently present. A fetal echocardiography showed normal cardiac activity, with a thoracic diameter well below the third percentile, in addition to the presence of a mild to moderate tricuspid regurgitation. The small thoracic diameter suggested a high probability of severe pulmonary hypoplasia.
A history of Zika virus infection was suspected when the mother was 20 weeks and 4 days pregnant due to clinical manifestations described as arthralgia during 10 days, moderate fever, and erythematous exanthema spots on the body. Real-time polymerase chain reaction (PCR) for Zika virus could not be performed.
At 39 weeks and 3 days of gestational age, the patient delivered a singleton male live newborn with facial malformation and very short limbs. The Apgar score was 1 for the 1st minute of life, and 0 for the 5th minute. The newborn died due to cardiorespiratory arrest 15 minutes after birth. A post-mortem examination of the baby was performed after we obtained the parents' authorization, and it included skeletal X-ray scans. Genetic molecular tests were not performed (Fig. 2).
(a) Patient ectoscopy: very short and deformed long bones; excess and wrinkled skin on a deformed face and neck; (b) non-ossification of the skull with transparent meninges; (c) extremely small thoracic cage; (d) typical babygram X-ray scan of a patient with a case of osteogenesis imperfecta type II.
The anatomopathological study revealed a deformed neonate boy measuring 38 cm (below the 3rd percentile) and weighing 1,800 g (below the 3rd percentile), with extreme shortening of all members. The skull was very soft, with no cranium ossification, and visualization of the meninges was evident. A narrow thoracic cage was present, and the shortening and deformities of the limbs were significant. The internal examination did not reveal any specific findings, except an important pulmonary hypoplasia (Fig. 2).
The radiographic images of the skeleton showed extreme shortening and deformity of the long bones with severe loss of ossification, especially on the skull. The bones, in general, presented an abnormal morphology, with “crumpled” humeri and femora, abnormally shaped ribs containing numerous fractures (“pearl appearance”), and a vertebral column with flattened vertebrae (platyspondylia) (Fig. 2).
Based on the clinical and radiological evidences, the patient was diagnosed as having OI type IIA (lethal form) (Figs. 1, 2, 3).
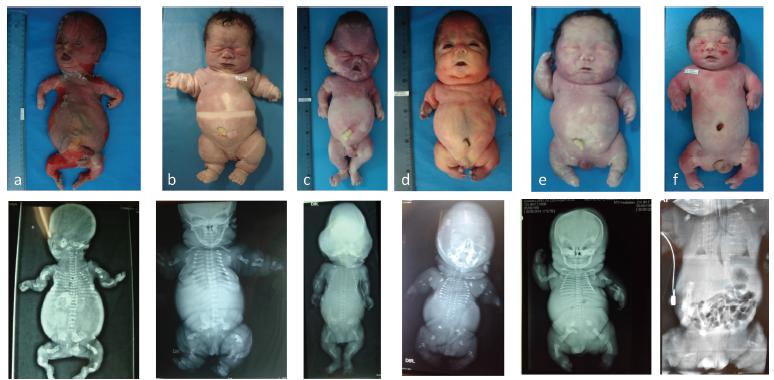
Most common types of lethal skeletal dysplasias: (a) osteogenesis imperfecta type IIA; (b) thanatophoric dysplasia type I; (c) thanatophoric dysplasia type II (observe the “cloverleaf” skull); (d) achondrogenesis type I; (e) fibrochondrogenesis; (f) hypophosphatasia.
Discussion
The lethal forms of SD represent a group of genetic disorders that are clinically and genetically heterogeneous, and whose cardinal manifestations are observed in the perinatal period with severe and prominent phenotypic features. The majority of deaths result from respiratory insufficiency due to pulmonary hypoplasia, with 23% of stillbirths and 32% of babies not surviving the first week of life.11. Camera G, Mastroiacovo P. Birth prevalence of skeletal dysplasias in the Italian Multicentric Monitoring System for Birth Defects. Prog Clin Biol Res 1982;104:441-449 44. Orioli IM, Castilla EE, Barbosa-Neto JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet 1986;23(04):328-332 In a clinical routine basis, the diagnosis of lethal SD occurs more frequently in the second trimester of gestation, through ultrasound findings (85%) and changes related to bone mineral density, including pathological fractures, growth deficiency, rib abnormalities, bowing or shortening of the long bones, and abnormal skull ossification, can be observed.1010. Krakow D, Williams J III, Poehl M, Rimoin DL, Platt LD. Use of three-dimensional ultrasound imaging in the diagnosis of prenatal- onset skeletal dysplasias. Ultrasound Obstet Gynecol 2003; 21(05):467-472 However, the ultrasonography findings do not always point to a specific SD, which may lead to an imprecise diagnosis, uncertainties and high expectations from the healthcare professionals and parents. In addition, due to the low incidence of lethal SDs, the presence of variable phenotypes, overlapping features and the lack of a positive family history, it is difficult to achieve a specific etiological diagnosis and, therefore, a clinical prognosis may be uncertain to access.
In the presence of an ultrasound finding indicative of a possible lethal SD, complementary tests can be of clinical relevance to document each case, such as three-dimensional (3D) ultrasound, magnetic resonance imaging with 3D reconstruction, and invasive methods for collecting material for molecular investigation through DNA extraction from the amniotic fluid or cordocentesis.1111. RuanoR,MolhoM,RoumeJ,VilleY. Prenatal diagnosisof fetal skeletal dysplasias by combining two-dimensional and three-dimensional ultrasound and intrauterine three-dimensional helical computer tomography. Ultrasound Obstet Gynecol 2004;24(02):134-140 In spite of being useful and of facilitating the visualization of the anatomical structures, there are exams that might be harmful to the fetus on account of the exposure to radiation, such as computed tomography, and others that present limitations in their interpretation due to the overlapping of fetal and maternal structures, such as the X-ray. However, the latter plays an extremely important role in the definition of phenotypic features in the post-natal period, especially when an SD is suspected. In addition to radiography, autopsy and collecting material for molecular investigation through fetal DNA analysis are ideal. Even so, in the absence of confirmatory genetic tests, a range of differential diagnoses of lethal SDs must be considered based on the clinical and radiological findings (Fig. 3).
The most common lethal SDs are thanatophoric dysplasia, OI type II, and the achondrogenesis group, which comprise 40 to 60% of the cases (Fig. 3). In a study conducted from 2010 to 2014, patients with a suspicion of SD were registered through the epidemiological birth defects program called Latin American Collaborative Study of Congenital Malformations (ECLAMC, in the Portuguese acronym).1212. Castilla EE, Orioli IM. ECLAMC: the Latin-American collaborative study of congenital malformations. Community Genet 2004; 7(2-3):76-94 The clinical and radiological examinations, as well as photographs, were evaluated when available based on a methodology with modifications proposed by Barbosa-Buck et al.1313. Barbosa-Buck CO, Orioli IM, da Graça Dutra M, Lopez-Camelo J, Castilla EE, Cavalcanti DP. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet A 2012;158A(05): 1038-1045 Three different diagnostic evidence levels (DELs) were established: DEL-1 (good quality X-rays and/or positive genotype, and/or follow-up clinical information defining the diagnosis); DEL-2 (satisfactory description of the clinical and radiological examinations to establish one or more probable diagnoses); and DEL-3- (clinical data and images with just enough quality to classify them as SDs).
Among 5,460 births, 1,452 newborns (26.6%) showed birth defects (2010–2014 ECLAMC survey, National Institute Fernandes Figueira). Among them, 32 babies (2%) had a suspicion of SD. The ultrasonographic diagnosis was suggestive in 93.7% of the pregnancies, and lethality during the prenatal period occurred in 81.2% of cases. In 50% of them, lethality was suspected after gestational ultrasounds. Out of 18 cases classified as DEL-1, 10 were of thanatophoric dysplasia (31.2%), 4 were of OI type II (12.5%) and there were 4 other cases each of diastrophic dysplasia, Verma-Naumoff syndrome, achondrogenesis and fibrochondrogenesis (Table 1).1414. Toriello HV, Meck JM. Professional Practice and Guidelines Committee. Statement on guidance for genetic counseling in advanced paternal age. Genet Med 2008;10(06):457-460 1515. Jones KL, Jones MC, Del Campo M. Smith's recognizable patterns of humanmalformation. 7thed. Philadelphia: Elsevier Saunders; 2013 Among the DEL-2 group, 6 cases had the following preliminary diagnosis: fibrochondrogenesis or Schneckenbecken dysplasia; opsismodysplasia; diastrophic dysplasia group (atelosteogenesis); hypochondroplasia; hypochondrogenesis; and campomelic dysplasia (OMIM, 2016).1616. OMIM(r) Online Mendelian Inheritance in Man [Internet]. Baltimore: McKusick-Nathans Institute of Genetic Medicine/Johns Hopkins University; 2016 [cited 2017 Jan 12]. Available from: https://omim.org/
Clinical, radiological and genetic characteristics in different lethal skeletal dysplasias1414. Toriello HV, Meck JM. Professional Practice and Guidelines Committee. Statement on guidance for genetic counseling in advanced paternal age. Genet Med 2008;10(06):457-460 1515. Jones KL, Jones MC, Del Campo M. Smith's recognizable patterns of humanmalformation. 7thed. Philadelphia: Elsevier Saunders; 2013
Currently, with the increasing advance in molecular diagnosis, diagnostic hypotheses can be precisely confirmed or excluded, thereby improving the accuracy of the genetic counselling. Hence, the importance of detailed clinical records of SD cases, including clinical and epidemiological data, radiographs, photographs, and storage of biological material (paraffin block-embedded tissue for DNA extraction, for example) is essential. Gene sequencing or exome sequencing for a panel of genes related to SD are now available.1717. Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015;167A(12):2869-2892
Once an experienced clinician in lethal SDs confirms the diagnosis, genetic counselling can be properly offered to the families.1818. Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med 2009;11(02): 127-133 In an autosomal dominant inheritance disease, the risk of recurrence for an affected parent can be as high as 50%; for parents who are not affected, the risk is negligible, as in OI type II or in the thanatophoric dysplasia group, unless there is the possibility of occurrence of germinal mosaicism.1919. Comstock JM, Putnam AR, Sangle N, Lowichik A, Rose NC, Opitz JM. Recurrence ofachondrogenesis type 2in sibs:Additional evidencefor germline mosaicism. Am J Med Genet A 2010;152A(07):1822-1824 When an autosomal recessive inheritance condition is established, such as hypophosphatasia or achondrogenesis, the parents are necessarily carriers, thus a 25% chance of recurrence may occur.
In the study by Barbosa-Buck et al,1313. Barbosa-Buck CO, Orioli IM, da Graça Dutra M, Lopez-Camelo J, Castilla EE, Cavalcanti DP. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet A 2012;158A(05): 1038-1045 the association of advanced paternal age with de novo SD cases was shown, especially in the thanatophoric dysplasia group. In the presence of advanced paternal age, there is a higher risk of occurrence of new mutations (de novo) per generation compared with advanced maternal age, especially in men, due to the large number of cell divisions during spermatogenesis.1414. Toriello HV, Meck JM. Professional Practice and Guidelines Committee. Statement on guidance for genetic counseling in advanced paternal age. Genet Med 2008;10(06):457-460
If the diagnosis of an SD is certain, it is essential to assess whether it is classified as lethal, since we should instruct parents as to the severity of the condition. In the present case, the diagnosis of OI type IIA was established at the postnatal period, due to radiological features consistent with the disease, such as extreme osteoporosis, presence of multiple fractures, “crumpled long bones,” absence of calcification of the skull, and blue sclera. Osteogenesis imperfecta type II, the most severe phenotype, has an incidence of ∼ 1 to 2 cases per 100,000 live births,2020. van Dijk FS, Cobben JM, Kariminejad A, et al. Osteogenesis imperfecta: a review with clinical examples. Mol Syndromol 2011;2(01):1-20 and its effects can already be observed in the uterus. Based on subtle radiographic differences, Sillence et al2121. Sillence DO, BarlowKK, Garber AP, Hall JG, Rimoin DL. Osteogenesis imperfecta type II delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 1984;17(02):407-423 subdivided the OI type II disorder into three further groups. Type IIA is characterized by short, broad crumpled femora and continuously beaded ribs; type IIB, by short, broad crumpled femora, but normal ribs or ribs with incomplete beading; and type IIC, by long, thin, inadequately modeled, rectangular long bones with multiple fractures, and thin, beaded ribs.1515. Jones KL, Jones MC, Del Campo M. Smith's recognizable patterns of humanmalformation. 7thed. Philadelphia: Elsevier Saunders; 2013
The newborns with lethal SDs survive during a few days after birth, and rarely survive for more than one year; the treatment involves intensive support and ventilatory assistance. More than 60% of affected newborns die on the first day of life, and 80% die within a week.88. Basel D, Steiner RD. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet Med 2009;11(06):375-385 In most cases, death usually occurs from respiratory failure related to severe pulmonary hypoplasia, rib fractures or unstable thoracic cage, but it can also be caused by pneumonia, hemorrhages in the central nervous system, and associated malformations. The treatment should focus on the relief of the symptoms and on support. The importance of medical assistance aiming at the quality of life of the baby through a palliative treatment to relieve the pain by means of potent analgesics should be emphasized. As the majority of newborns die in the perinatal period, rapid and effective assistance is essential for the patients' comfort, with minimal handling due to the risk of fractures when it is the case, such as in this report of OI type IIA.
With the same importance, we must emphasize the relevance of medical documentation as much as radiological plates and photographs, especially in cases in which no etiological diagnosis has been established with certainty. The anatomopathological study is recommended in cases in which there is presence or suspicion of associated congenital malformations, such as polydactyly. Findings of internal malformations suggested by gestational ultrasound should be confirmed, and they contribute to the differential diagnosis of the lethal cases associated with congenital malformations, such as the short-rib polydactyly group1515. Jones KL, Jones MC, Del Campo M. Smith's recognizable patterns of humanmalformation. 7thed. Philadelphia: Elsevier Saunders; 2013 (Table 1).
The prognosis of a lethal SD, due to the severity of the condition, is quite limited. The physician must be used to talking to parents about the possibility of a lethal case, which itself requires great sensitivity and empathy. The use of simple and accessible language facilitates the decision-making in the management and treatment of such cases. As there may be implications in the course of pregnancy and during the postnatal care, the parents should receive multidisciplinary support and adequate guidance regarding the continuation of pregnancy, the risks of recurrence for new pregnancies and the postnatal care, respecting the culture, religion and the laws of each community.
Clinical management and decision-making in cases in which a lethal disease is suspected in the prenatal period, such as a lethal SD, demands a multidisciplinary approach coordinated by an experienced physician. Firstly, a “clinical descriptive” approach, instead of a “need for diagnosis” approach, is mostly recommended. Secondly, all efforts in collecting good quality documentation, including X-ray plates and photographic material, are essential. Thirdly, biological samples (amniotic fluid, blood, paraffin block-embedded tissue) for DNA extraction and posterior gene sequencing (exome panel or Sanger gene sequencing) should be stored; and, lastly, autopsy, including post-mortem X-ray plates, once the multiple congenital malformations suspected should be pursued. The genetic counselling for a case of lethal SD will be enormously enriched with the precise knowledge of the recurrence risks if each of these recommendations is fulfilled.
Acknowledgments
This case report and photographic documentation were approved for publication by the Ethical Committee Board of one of our institutions, under protocol numbers CAAE 0022.0.008.000-08 (osteogenesis imperfecta) and CAEE 59488716.1.1001.5269 (ECLAMC).
References
-
1Camera G, Mastroiacovo P. Birth prevalence of skeletal dysplasias in the Italian Multicentric Monitoring System for Birth Defects. Prog Clin Biol Res 1982;104:441-449
-
2Rasmussen SA, Bieber FR, Benacerraf BR, Lachman RS, Rimoin DL, Holmes LB. Epidemiology of osteochondrodysplasias: changing trends due to advances in prenatal diagnosis. Am J Med Genet 1996;61(01):49-58
-
3Andersen PE Jr. Prevalence of lethal osteochondrodysplasias in Denmark. Am J Med Genet 1989;32(04):484-489
-
4Orioli IM, Castilla EE, Barbosa-Neto JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet 1986;23(04):328-332
-
5Krakow D, Alanay Y, Rimoin LP, et al. Evaluation of prenatal-onset osteochondrodysplasias by ultrasonography: a retrospective and prospective analysis. AmJMed Genet A2008;146A(15):1917-1924
-
6Tretter AE, Saunders RC, Meyers CM, et al. Antenatal diagnosis of lethal skeletal dysplasias. Am J Med Genet 1998;75(05):518-522
-
7Källén B, Knudsen LB, Mutchinick O, et al. Monitoring dominant germ cell mutations using skeletal dysplasias registered in malformation registries: an international feasibility study. Int J Epidemiol 1993;22(01):107-115
-
8Basel D, Steiner RD. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet Med 2009;11(06):375-385
-
9Huber MA. Osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;103(03):314-320
-
10Krakow D, Williams J III, Poehl M, Rimoin DL, Platt LD. Use of three-dimensional ultrasound imaging in the diagnosis of prenatal- onset skeletal dysplasias. Ultrasound Obstet Gynecol 2003; 21(05):467-472
-
11RuanoR,MolhoM,RoumeJ,VilleY. Prenatal diagnosisof fetal skeletal dysplasias by combining two-dimensional and three-dimensional ultrasound and intrauterine three-dimensional helical computer tomography. Ultrasound Obstet Gynecol 2004;24(02):134-140
-
12Castilla EE, Orioli IM. ECLAMC: the Latin-American collaborative study of congenital malformations. Community Genet 2004; 7(2-3):76-94
-
13Barbosa-Buck CO, Orioli IM, da Graça Dutra M, Lopez-Camelo J, Castilla EE, Cavalcanti DP. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet A 2012;158A(05): 1038-1045
-
14Toriello HV, Meck JM. Professional Practice and Guidelines Committee. Statement on guidance for genetic counseling in advanced paternal age. Genet Med 2008;10(06):457-460
-
15Jones KL, Jones MC, Del Campo M. Smith's recognizable patterns of humanmalformation. 7thed. Philadelphia: Elsevier Saunders; 2013
-
16OMIM(r) Online Mendelian Inheritance in Man [Internet]. Baltimore: McKusick-Nathans Institute of Genetic Medicine/Johns Hopkins University; 2016 [cited 2017 Jan 12]. Available from: https://omim.org/
-
17Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015;167A(12):2869-2892
-
18Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med 2009;11(02): 127-133
-
19Comstock JM, Putnam AR, Sangle N, Lowichik A, Rose NC, Opitz JM. Recurrence ofachondrogenesis type 2in sibs:Additional evidencefor germline mosaicism. Am J Med Genet A 2010;152A(07):1822-1824
-
20van Dijk FS, Cobben JM, Kariminejad A, et al. Osteogenesis imperfecta: a review with clinical examples. Mol Syndromol 2011;2(01):1-20
-
21Sillence DO, BarlowKK, Garber AP, Hall JG, Rimoin DL. Osteogenesis imperfecta type II delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 1984;17(02):407-423
Publication Dates
-
Publication in this collection
Oct 2017
History
-
Received
13 Dec 2016 -
Accepted
04 Apr 2017