Abstract
Schwann cell disturbance followed by segmental demyelination in the peripheral nervous system occurs in diabetic patients. Since Schwann cell and oligodendrocyte remyelination in the central nervous system is a well-known event in the ethidium bromide (EB) demyelinating model, the aim of this investigation was to determine the behavior of both cell types after local EB injection into the brainstem of streptozotocin diabetic rats. Adult male Wistar rats received a single intravenous injection of streptozotocin (50 mg/kg) and were submitted 10 days later to a single injection of 10 µL 0.1% (w/v) EB or 0.9% saline solution into the cisterna pontis. Ten microliters of 0.1% EB was also injected into non-diabetic rats. The animals were anesthetized and perfused through the heart 7 to 31 days after EB or saline injection and brainstem sections were collected and processed for light and transmission electron microscopy. The final balance of myelin repair in diabetic and non-diabetic rats at 31 days was compared using a semi-quantitative method. Diabetic rats presented delayed macrophage activity and lesser remyelination compared to non-diabetic rats. Although oligodendrocytes were the major remyelinating cells in the brainstem, Schwann cells invaded EB-induced lesions, first appearing at 11 days in non-diabetic rats and by 15 days in diabetic rats. Results indicate that short-term streptozotocin-induced diabetes hindered both oligodendrocyte and Schwann cell remyelination (mean remyelination scores of 2.57 ± 0.77 for oligodendrocytes and 0.67 ± 0.5 for Schwann cells) compared to non-diabetic rats (3.27 ± 0.85 and 1.38 ± 0.81, respectively).
Central nervous system; Diabetes mellitus; Ethidium bromide; Remyelination; Schwann cell; Oligodendrocyte
Braz J Med Biol Res, May 2006, Volume 39(5) 637-646
Delayed Schwann cell and oligodendrocyte remyelination after ethidium bromide injection in the brainstem of Wistar rats submitted to streptozotocin diabetogenic treatment
 Correspondence and Footnotes
Correspondence and Footnotes
 E.F. Bondan1, M.A. Lallo1,2, A.H. Trigueiro1, C.P. Ribeiro3, I.L. Sinhorini3 and D.L. Graça4
E.F. Bondan1, M.A. Lallo1,2, A.H. Trigueiro1, C.P. Ribeiro3, I.L. Sinhorini3 and D.L. Graça4
1Mestrado em Ciências da Reabilitação Neuromotora, Universidade Bandeirante de São Paulo, São Paulo, SP, Brasil
2Departamento de Medicina Veterinária, Universidade Paulista, São Paulo, SP, Brasil
3Departamento de Patologia Veterinária, Faculdade de Medicina Veterinária e Zootecnia, Universidade de São Paulo, São Paulo, SP, Brasil
4Departamento de Patologia, Faculdade de Medicina Veterinária, Universidade Federal de Santa Maria, Santa Maria, RS, Brasil
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Schwann cell disturbance followed by segmental demyelination in the peripheral nervous system occurs in diabetic patients. Since Schwann cell and oligodendrocyte remyelination in the central nervous system is a well-known event in the ethidium bromide (EB) demyelinating model, the aim of this investigation was to determine the behavior of both cell types after local EB injection into the brainstem of streptozotocin diabetic rats. Adult male Wistar rats received a single intravenous injection of streptozotocin (50 mg/kg) and were submitted 10 days later to a single injection of 10 µL 0.1% (w/v) EB or 0.9% saline solution into the cisterna pontis. Ten microliters of 0.1% EB was also injected into non-diabetic rats. The animals were anesthetized and perfused through the heart 7 to 31 days after EB or saline injection and brainstem sections were collected and processed for light and transmission electron microscopy. The final balance of myelin repair in diabetic and non-diabetic rats at 31 days was compared using a semi-quantitative method. Diabetic rats presented delayed macrophage activity and lesser remyelination compared to non-diabetic rats. Although oligodendrocytes were the major remyelinating cells in the brainstem, Schwann cells invaded EB-induced lesions, first appearing at 11 days in non-diabetic rats and by 15 days in diabetic rats. Results indicate that short-term streptozotocin-induced diabetes hindered both oligodendrocyte and Schwann cell remyelination (mean remyelination scores of 2.57 ± 0.77 for oligodendrocytes and 0.67 ± 0.5 for Schwann cells) compared to non-diabetic rats (3.27 ± 0.85 and 1.38 ± 0.81, respectively).
Key words: Central nervous system, Diabetes mellitus, Ethidium bromide, Remyelination, Schwann cell, Oligodendrocyte
Introduction
Segmental demyelination is a prominent feature of the symmetric polyneuropathy of diabetic patients (1,2), probably due to Schwann cell disturbance. The mechanism of disturbance may be related to the fact that Schwann cells are a primary target of polyol pathway abnormalities strongly linked to the hyperglycemia found in diabetes mellitus (3). Schwann cell invasion and contribution to myelin repair in the central nervous system (CNS) are usually seen in the ethidium bromide (EB) demyelinating model (4-14), where there is early astrocyte disappearance, with both glia limitans and blood-brain barrier disruption. Oligodendroglial damage induced by EB produces primary demyelinating lesions at the site of injection, with subsequent remyelination performed by surviving oligodendrocytes and by invasive Schwann cells (4-6,9,12-14).
Since Schwann cell and oligodendrocyte remyelination in the CNS is a well-known event in demyelinating diseases such as multiple sclerosis (15-17) as well as in the EB gliotoxic model, the aim of the present investigation was to determine the behavior of these myelin-repairing cells after local EB injection in the brainstem of rats submitted to the diabetogenic model of streptozotocin.
Material and Methods
Sixty-nine adult male Wistar rats were used. Of these, 42 received a single injection of 50 mg/kg streptozotocin in 10 mM sodium citrate buffer, pH 4.5, into the tail vein after 12 h of fasting. Ten days later blood glucose levels were measured and animals with levels of 200 mg/dL or more were considered to be diabetic. At that time some rats were submitted to a injection of 10 µL 0.1% (w/v) EB (group I, N = 27) or 0.9% saline (group II, N = 10) into the cisterna pontis, while others did not receive intracisternal injection and were used as histologic controls (group III, N = 5). The EB- or saline-injected rats were anesthetized with ketamine and xylazine (5:1; 0.1 mL/100 g) and a burr-hole was made on the right side of the skull, 8 mm rostral to the fronto-parietal suture. Injections were performed freehand using a Hamilton syringe fitted with a 35o angled polished 26-gauge needle into the cisterna pontis, an enlarged subarachnoid space below the ventral surface on the pons. Non-diabetic rats (group IV, N = 27) received 10 µL 0.1% EB solution. Body weight and blood glucose levels (Dextrostix, Ames Laboratory, Ames, IA, USA) were recorded on three occasions, at the time of streptozotocin injection, 10 days later and at the time of sacrifice. Rats were kept under controled light conditions (12-h light-dark cycle) and water and food were given ad libitum during the experimental period.
At different times after EB or saline injection, the rats were anesthetized and submitted to intracardiac perfusion with 4% (v/v) glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.4. Three animals each from group I and 3 from group IV were perfused at 7, 11, 15, and 21 days after intracisternal injection, and 15 rats from each of these groups were perfused at 31 days. Two animals from group II and 1 from group III were perfused at 7, 11, 15, 21, and 31 days. Thin sections of the brainstem (pons, medulla oblongata and mesencephalon) were collected and post-fixed in 0.1% osmium tetroxide, dehydrated with graded acetones and embedded in Araldite 502 resin, following transitional stages in acetone. Thick sections were stained with 0.25% alkaline toluidine blue. Selected areas were trimmed and thin sections were stained with 2% uranyl and lead acetate and viewed in a Philips EM-201 transmission electron microscope.
The final balance of myelin repair was compared between diabetic and non-diabetic rats using the semi-quantitative method developed by Blakemore and Crang (18) and Gilson and Blakemore (19) to document the extent and nature of remyelination in gliotoxic lesions. Two semithin sections from each animal from groups I and IV obtained 31 days after EB injection were examined for the presence of axons remyelinated by oligodendrocytes and Schwann cells, as well as for demyelinated axons. Remyelination by either Schwann cells or oligodendrocytes was identified using previously described morphological criteria (6). Since the thickness of normal myelin sheaths bears a constant relationship to the axons they surround, repaired myelin sheaths can be identified because they are thinner than normal for the axons they cover. At the ultrastructural level, myelin sheaths produced by oligodendrocytes have characteristic appearance and periodicity. Schwann cells are not present within the white matter of the CNS. Schwann cells relate to single internodes of myelin, and, as peripheral myelin has a different chemical composition from central myelin, this results in a different appearance between Schwann cell and oligodendrocyte myelin following staining with toluidine blue. At the ultrastructural level, Schwann cell myelin has a periodicity that differs from myelin produced by oligodendrocytes. Furthermore, the outer surface of myelin formed by Schwann cells is covered by a basement membrane and Schwann cell nuclei are often seen lying next to their myelin sheaths.
The proportion of each was estimated on a scale ranging from 0 to 5. A lesion in which all axons were remyelinated by oligodendrocytes would have an oligodendrocyte (O) score of 5, a Schwann cell (S) score of 0 and a demyelination (D) score of 0. If 40% of the demyelinated axons were remyelinated by oligodendrocytes and 40% by Schwann cells with the remaining axons being demyelinated, then the lesion was assigned a score of O-2, S-2, D-1. To compare remyelination scores from diabetic and non-diabetic rats 31 days after EB injection, a lesion repair profile of O versus S remyelination was made, providing an adequate graphic representation of each group. The mean O and S scores ± 2 SEM enclosed a domain which represented an average of repair. Non-overlapping domains in these graphic representations indicated significantly different results.
Results
Non-diabetic rats treated with ethidium bromide (group IV)
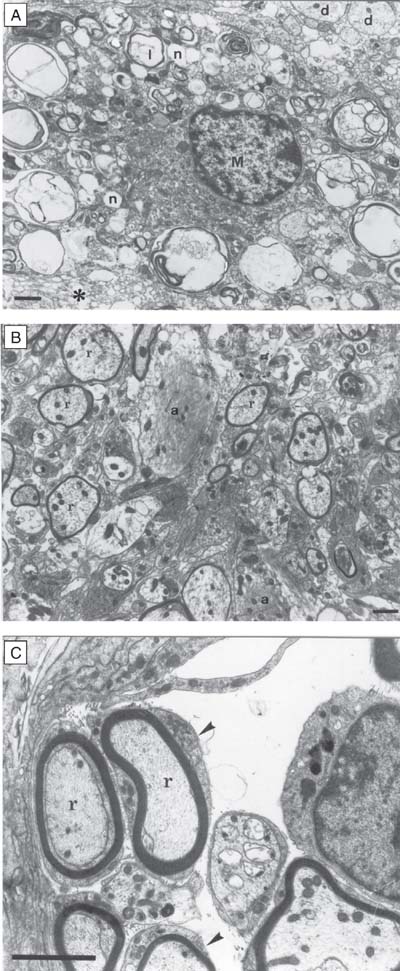
The EB-induced lesions in non-diabetic rats were similar to those described in the brainstem by Pereira et al. (14) and by Bondan et al. (6). They were characterized by demyelinating lesions on the ventral surface of the pons and mesencephalon, containing in the central area phagocytic cells, some myelin-derived membranes in a distended extracellular space as well as demyelinated axons (Figure 1A).
At the periphery, the presence of oligodendrocytes and Schwann cells was noted at 11 days after EB administration, the latter occurring in areas of an enlarged extracellular space devoid of astrocytic processes, notably round blood vessels and subpial areas. Oligodendrocytes showed an incipient but preponderant remyelination (Figure 1B), though Schwann cells also appeared to contribute to myelin repair (Figure 1C). Remyelination produced by both cells increased with time, but oligodendroglia continued to prevail in the brainstem myelin repair, although sheaths formed by Schwann cells were thicker than those produced by oligodendrocytes during the same period. Lymphocytes were observed 7 to 31 days after EB injection, contacting phagocytic cells and myelin debris. Infiltrating pial cells were also seen within the lesions, appearing as compact and monotypic nests or in diffuse arrangements within the lesions.
Group IV. A, 7 days after ethidium bromide injection. A macrophage (M) containing myelin at different stages of degradation, including lamellae (l) and neutral fat droplets (n) in an area of enlarged extracellular space filled with myelin debris (asterisk) and demyelinated axons (d). Bar = 1 µm. B, 15 days after ethidium bromide injection. Oligodendrocyte-remyelinated axons (r) associated with hyperthrophic astrocytic processes (a). Bar = 1 µm. C, 15 days after ethidium bromide injection. Axons (r) remyelinated by Schwann cells. Note that portions of Schwann cell cytoplasm (arrowheads) surroundthese axons. Bar = 2 µm.
Clinical observations of diabetic rats
All rats submitted to the streptozotocin injection presented hyperglycemia (levels from 200 to 450 mg/dL) on the 10th day and on the day of perfusion. During the experimental period they developed characteristic polyuria, polydipsia and body weight loss. Plasma glucose levels and body weight data are shown in Table 1.
Ethidium bromide-injected diabetic rats (group I)
By 7-11 days after induction of the gliotoxic lesion, two distinct areas were seen, a central one and a peripheral one. The center of the lesion was expanded and filled with huge amounts of myelin debris among foamy macrophages, demyelinated axons (clumped or isolated) and occasional degenerating axons (Figure 2A). No astrocytic processes were found in this site. Hemorrhagic areas were observed from 7 to 15 days post-injection and lymphocytes were seen within the neuropil and in perivascular locations. By 11 days, some cells with morphological features of oligodendrocytes were seen around the central area, but without any indication of remyelinating activity.
The most prominent feature of the 15th day after EB injection was the initial association of naked axons and remyelinating cells at peripheral locations. Schwann cells were associated with one or multiple demyelinated axons or were already forming thin myelin lamellae around single axons in astrocyte-free areas (Figure 2B). In contrast, oligodendrocytes began to form slight myelin sheaths in areas completely or partially filled with astrocytic processes (Figure 2C). In addition to peripheral astrogliosis, pial cell infiltration was also noted from 15 to 31 days after the gliotoxic injection and, although rare, signs of axonal degeneration persisted until the 15th day after injection.
By 21 and 31 days, it became evident that diabetic rats presented a considerable delay in the repair process compared to non-diabetic animals, with greater amounts of myelin-derived membranes in the central area and a lesser extent of peripheral remyelination by both oligodendrocytes and Schwann cells. In diabetic rats, a greater proportion of axons persisted without myelin and the remyelinated ones clearly presented thinner myelin sheaths (Figure 2D) in relation to non-diabetic rats from group IV.
Group I. A, 7 days after ethidium bromide injection. Demyelinated axons (d) in an area containing large amounts of myelin-derived membranes (asterisks), phagocytic cells (M) and even a degenerating axon (arrow). Bar = 1 µm. B, 15 days after ethidium bromide injection. A Schwann cell (S) surrounds a naked axon near some oligodendrocyte-remyelinated axons (r). Bar = 5 µm. C, 15 days after ethidium bromide injection. An oligodendrocyte (O) is seen next to thinly remyelinated axons (r). Bar = 1 µm. D, 21 days after ethidium bromide injection. Axons present initial deposition of few myelin lamellae (arrowheads) of oligodendroglial origin. Bar = 0.5 µm.
Rats injected with saline solution (group II)
Mild lesions circumscribed to the pons and along the needle track were detected 7 to 15 days post-injection due to the surgical procedure. Ultrastructural analysis of the lesions showed a light and focal expansion of the extracellular space containing some loose lamellae and myelin debris as well as phagocytic macrophages. Few fibers showed periaxonal edema and signs of degeneration, but there was no evidence of primary demyelination or loss of neuroglia.
Score comparison between groups I and IV at 31 days
Group I (diabetic rats) and IV (non-diabetic rats) were compared by scanning semithin sections 31 days after EB injection, with documentation of the remyelination balance and construction of the corresponding diagrams, according to the semi-quantitative method developed by Blakemore and Crang (18) and Gilson and Blakemore (19). Scores are presented in Table 2 and the respective diagrams in Figure 3.
Diagrams of oligodendrocyte (O) versus Schwann cell (S) remyelination scores in non-diabetic (A) and diabetic (B) rats 31 days post-lesioning. The areas within circles represent mean ± 2 SEM and are considered as domains that represent an average of repair. Non-overlapping repair domains indicate significantly different results. The majority of axons are remyelinated by oligodendrocytes.
Remyelination scores 31 days after ethidium bromide injection in groups I (diabetic rats) and IV (non-diabetic rats).
Discussion
Two major features were identified in the EB demyelinating model in Wistar rats, i.e., the degenerative features during the first phase post-injection of the gliotoxin, and the reconstructive events related to the repair of the induced lesion.
In the first phase, identified in the present study by the morphological findings at 7 and 11 days post-injection, glial cells completed their degenerative changes, with consequent demyelination followed by macrophage removal of cellular debris. In the second, from the 15th to the 31st day after injection, the dynamics of the remyelinating process was observed, with astrogliosis present or not in specific areas, as well as lack of remyelination in other sites of the lesion.
These events have been exhaustively discussed in different investigations involving the gliotoxic agent EB in the brainstem (6-8,12-14) or in the spinal cord (11,20) of rats.
Although similar cellular phenomena related to the EB model have appeared in streptozotocin-diabetic rats, it was noted that the temporal pattern of repair was somehow different from that seen in non-diabetic rats, caused by the delay in the scavenging activity of macrophages and in the subsequent remyelinating process in the diabetic group. This pattern resembled the one observed in rats immunosuppressed with dexamethasone (5,9) or cyclophosphamide (6), in which incipient and limited oligodendrocyte remyelination, predominance of naked axons, delayed neovascularization, and scanty lymphocyte infiltration were observed.
Delay in macrophage function in these immunosuppressed animals was indicated by the accumulation of huge amounts of myelin-derived membranes in the center of the lesion, without any interference of either astrocytic reaction following EB injection, or in the invasive and remyelinating activity of Schwann cells in the brainstem (5,6,9).
When these results are compared with those previously described in the EB model using the same dose (10 µL x 0.1% EB), animal species, breed and age (adult Wistar rats), and site of injection (cisterna pontis), the following differences were found in the diabetic group regarding Schwann cell infiltration within the induced lesions. First, these cells were only identified from the 15th day post-injection and not from the 11th day as seen in the non-diabetic rats of the present investigation and as also reported by Pereira et al. (14). Second, Schwann cell remyelinating activity was apparently delayed if we consider the remyelination seen in the non-diabetic group (group IV) and also described by Pereira et al. (14) and Bondan et al. (4,6,9), although the distribution of Schwann cells was rather the same - in areas of expanded extracellular space, devoid of astrocytic processes, notably around blood vessels, and in subpial areas. Finally, we did not observe simultaneous myelin deposition in 2 or more internodes by the same Schwann cell, or redundant myelin loops of peripheral origin, as opposed to that observed in group IV and also in normal rats (4,6).
In the brainstem, Yajima and Suzuki (13) and Pereira et al. (14) first detected Schwann cells in the EB-induced lesions at 10 and 11 days post-injection, respectively. In this context, it can be considered that the Schwann cell invasion found in the diabetic rats of the present study was a little delayed. The origin of these invasive Schwann cells remains uncertain, but it is proposed that, with the transitory disappearance of astrocytes, they may migrate from pial nerves or nerves associated to encephalic blood vessels, as well as from the roots of spinal or cranial nerves (10).
In the present study, there was no decrease in lymphocytic infiltration as observed in rats immunosuppressed with cyclophosphamide (6) or dexamethasone (5,9), a fact that may suggest that the diabetic state achieved by these animals was not sufficient to compromise their immunological function. However, there were indications that macrophage activity was disturbed based on the finding of huge amounts of myelin-derived membranes, suggesting that short-term diabetes mellitus mimics lesions of slow development induced by EB in the rat CNS and delays all the subsequent repair events (5,6,9,11). Furthermore, abnormal macrophage function has been described in diabetes mellitus (3,21) and may explain why clearance of cellular debris tends to be a longer-term process in group I.
No ultrastructural alteration commonly found in the peripheral nervous system of diabetic patients was seen in the invasive Schwann cells, such as onion-bulb formations around remyelinated axons and/or myelin splitting or ballooning of the already formed myelin sheaths (1,22).
Signs of axonal degeneration were minimal and were observed from 7 to 15 days after EB or saline injection in all groups (diabetic or not), attributed to the injection trauma and not to the diabetic status, when degeneration would be progressive in the subsequent periods. The lack of indications of axonal degeneration due to the diabetic condition at the subsequent time points after the 15th day agrees with Thomas (2), according to whom diabetogenic states of short duration are not sufficient to provoke morphologically detectable axonal alterations.
It is important to stress that the disturbance in insulin secretion induced by streptozotocin may not have lasted long enough to interfere with Schwann cell invasion and remyelination of the demyelinated CNS axons. What was seen was a delayed invasion of Schwann cells in the brainstem when compared with their invasive activity in non-diabetic rats.
Insulin and IGF-1 levels are decreased in experimental streptozotocin diabetes (23,24) and both are recognized as important stimulating factors for Schwann cells (25-27).
In cells that do not need insulin for glucose transport, such as Schwann cells, hyperglycemia usually causes intracellular glucose accumulation and increased polyol pathway activity by aldose reductase resulting in increased levels of sorbitol and fructose, a fact that may cause osmotic swelling and even cell death. Hyperglycemia may provoke a decrease in tissue levels of myo-inositol and this is hypothesized to cause a sequence of events leading to deficiency of sodium-potassium-ATPase activity. The deposition of these polyols diverts the cellular machinery from the formation of normal intermediate metabolites, altering the complex equilibrium between intracellular signaling networks and target enzymes (3).
It is known that polyol metabolism disturbance with increased concentrations of endoneural sorbitol and fructose is observed as early as 3 days after the injection of streptozotocin, with an increase in aldose reductase activity within 1 week (28,29). If this occurred in our study, it was not sufficient to induce detectable ultrastructural changes in the migrating Schwann cells.
The scant oligodendroglial remyelination observed in the present experiment could also be the result of a lack of important trophic factors for the proliferation and differentiation of surviving oligodendrocytes and/or of their progenitors, the bipotential O-2A cells. It is recognized, for instance, that IGF-1 and -2 stimulate the proliferation of these cells (30) and receptors for IGF-1 are expressed in astrocytes and oligodendrocytes present on the edges of demyelinating lesions (31).
It is important to confront the results of this short-term experiment with a new one in which the same EB toxic demyelination should be achieved in conditions of long-term diabetes mellitus, in order to verify if Schwann cells and even oligodendrocytes present any additional and cumulative damage from being exposed to chronic hyperglycemia and enduring accumulation of sorbitol and fructose.
With the semi-quantitative analysis (by comparing the domains of the mean scores for groups I and IV and their limits - ± 2 SEM - at 31 days), it is clear that short-term diabetes induced by streptozotocin in the present study caused, in general terms, a defective repair process in the EB-induced lesions, affecting both Schwann cell and oligodendroglial remyelination, with persistence of a large proportion of demyelinated axons.
Acknowledgments
The authors wish to thank Mrs. Shirley Meire da Silva (USP) for technical assistance.
Publication supported by FAPESP. Received May 19, 2005. Accepted January 16, 2006.
- 1. Mizisin AP, Shelton GD, Wagner S et al. (1998). Myelin splitting, Schwann cell injury and demyelination in feline diabetic neuropathy. Acta Neuropathologica, 95: 171-174.
- 2. Thomas PK (1984). Animal models of diabetic neuropathies. In: Dyck PJ, Thomas PK, Lambert EH, Bunge R (Editors), Peripheral Neuropathy W.B. Saunders, Philadelphia, PA, USA, 691-706.
- 3. Cotran RS, Kumar V & Collins SL (2000). Robbins - Pathologic Basis of Disease W.B. Saunders, Philadelphia, PA, USA.
- 4. Bondan EF, Lallo MA, Sinhorini IL et al. (1999). Schwann cells may express an oligodendrocyte-like remyelinating pattern following ethidium bromide injection in the rat brainstem. Microscopica Acta, 8: 707-708.
- 5. Bondan EF, Lallo MA, Sinhorini IL et al. (1999). Ultrastructural investigation on the brainstem remyelination after local ethidium bromide injection in rats immunosuppressed with dexamethasone. Microscopica Acta, 8: 709-710.
- 6. Bondan EF, Lallo MA, Sinhorini IL et al. (2000). The effect of cyclophosphamide on brainstem remyelination following local ethidium bromide injection in Wistar rats. Journal of Submicroscopic Cytology and Pathology, 32: 603-612.
- 7. Bondan EF, Lallo MA, Dagli ML et al. (2002). Blood-brain barrier breakdown following gliotoxic drug injection in the brainstem of Wistar rats. Arquivos de Neuro-Psiquiatria, 60: 582-589.
- 8. Bondan EF, Lallo MA, Dagli ML et al. (2003). Investigation into the astrocytic immunoreactivity to GFAP and vimentin in the brainstem of Wistar rats submitted to the ethidium bromide gliotoxic model. Arquivos de Neuro-Psiquiatria, 61: 642-649.
- 9. Bondan EF, Lallo MA, Baz EI et al. (2004). Ultrastructural study of the remyelinating process following local ethidium bromide injection in the brainstem of dexamethasone-immunosuppressed rats. Arquivos de Neuro-Psiquiatria, 62: 131-138.
- 10. Graça DL (1989). Desmielinização tóxica do sistema nervoso central. II. Aspectos biológicos das células de Schwann observados durante o processo de reparação do tecido. Arquivos de Neuro-Psiquiatria, 47: 268-273.
- 11. Graça DL & Blakemore WF (1986). Delayed remyelination in rat spinal cord following ethidium bromide injection. Neuropathology and Applied Neurobiology, 12: 593-605.
- 12. Reynolds R & Wilkin GP (1993). Cellular reaction to an acute demyelinating/remyelinating lesion of the rat brain stem: localisation of GD3 ganglioside immunoreactivity. Journal of Neuroscience Research, 36: 405-422.
- 13. Yajima K & Suzuki K (1979). Ultrastructural changes of oligodendroglia and myelin sheaths induced by ethidium bromide. Neuropathology and Applied Neurobiology, 5: 49-62.
- 14. Pereira LA, Dertkigil MS, Graca DL et al. (1998). Dynamics of remyelination in the brain of adult rats after exposure to ethidium bromide. Journal of Submicroscopic Cytology and Pathology, 30: 341-348.
- 15. Itoyama Y, Webster HD, Richardson Jr EP et al. (1983). Schwann cell remyelination of demyelinated axons in spinal cord multiple sclerosis lesions. Annals of Neurology, 14: 339-346.
- 16. Itoyama Y, Ohnishi A, Tateishi J et al. (1985). Spinal cord multiple sclerosis lesions in Japanese patients: Schwann cell remyelination occurs in areas that lack glial fibrillary acidic protein (GFAP). Acta Neuropathologica, 65: 217-223.
- 17. Ogata J & Feigin I (1975). Schwann cells and regenerated peripheral myelin in multiple sclerosis: an ultrastructural study. Neurology, 25: 713-716.
- 18. Blakemore WF & Crang AJ (1989). The relationship between type-1 astrocytes, Schwann cells and oligodendrocytes following transplantation of glial cell cultures into demyelinating lesions in the adult rat spinal cord. Journal of Neurocytology, 18: 519-528.
- 19. Gilson J & Blakemore WF (1993). Failure of remyelination in areas of demyelination produced in the spinal cord of old rats. Neuropathology and Applied Neurobiology, 19: 173-181.
- 20. Blakemore WF (1982). Ethidium bromide induced demyelination in the spinal cord of the cat. Neuropathology and Applied Neurobiology, 8: 365-375.
- 21. Geisler C, Almdal T, Bennedsen J et al. (1982). Monocyte functions in diabetes mellitus. Acta Pathologica Microbiologica et Immunologica Scandinavica, 90: 33-37.
- 22. Ballin RH & Thomas PK (1968). Hypertrophic changes in diabetic neuropathy. Acta Neuropathologica, 11: 93-102.
- 23. Crosby SR, Tsigos C, Anderton CD et al. (1992). Elevated plasma insulin-like growth factor binding protein-1 levels in type 1 (insulin-dependent) diabetic patients with peripheral neuropathy. Diabetologia, 35: 868-872.
- 24. Goldstein S, Sertich GJ, Levan KR et al. (1988). Nutrition and somatomedin. XIX. Molecular regulation of insulin-like growth factor-1 in streptozotocin-diabetic rats. Molecular Endocrinology, 2: 1093-1100.
- 25. Dubovy P & Svizenska I (1992). Migration of Schwann cells from the distal stump of the sciatic nerve 1 week after transection: the effects of insulin and cytosine arabinoside. Glia, 6: 281-288.
- 26. Jung-Testas I, Schumacher M, Robel P et al. (1994). Actions of steroid hormones- and growth factors on glial cells of the central and peripheral nervous system. Journal of Steroid Biochemistry and Molecular Biology, 48: 145-154.
- 27. Schumacher M, Jung-Testas I, Robel P et al. (1993). Insulin-like growth factor I: a mitogen for rat Schwann cells in the presence of elevated levels of cyclic AMP. Glia, 8: 232-240.
- 28. Varma SD & Kinoshita JH (1974). Sorbitol pathway in diabetic and galactosemic rat lens. Biochimica et Biophysica Acta, 338: 632-640.
- 29. Ward JD, Baker RW & Davis BH (1972). Effect of blood sugar control on the accumulation of sorbitol and fructose in nervous tissues. Diabetes, 21: 1173-1178.
- 30. Goldman JE (1992). Regulation of oligodendrocyte differentiation. Trends in Neurosciences, 15: 359-362.
- 31. Komoly S, Hudson LD, Webster HD et al. (1992). Insulin-like growth factor I gene expression is induced in astrocytes during experimental demyelination. Proceedings of the National Academy of Sciences, 89: 1894-1898.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
24 Apr 2006 -
Date of issue
May 2006
History
-
Received
19 May 2005 -
Accepted
16 Jan 2006