Abstract
Malignant hyperthermia (MH) is a pharmacogenetic disease triggered in susceptible individuals by the administration of volatile halogenated anesthetics and/or succinylcholine, leading to the development of a hypermetabolic crisis, which is caused by abnormal release of Ca2+ from the sarcoplasmic reticulum, through the Ca2+ release channel ryanodine receptor 1 (RyR1). Mutations in the RYR1 gene are associated with MH in the majority of susceptible families. Genetic screening of a 5-generation Brazilian family with a history of MH-related deaths and a previous MH diagnosis by the caffeine halothane contracture test (CHCT) in some individuals was performed using restriction and sequencing analysis. A novel missense mutation, Gly4935Ser, was found in an important functional and conserved locus of this gene, the transmembrane region of RyR1. In this family, 2 MH-susceptible individuals previously diagnosed with CHCT carry this novel mutation and another 24 not previously diagnosed members also carry it. However, this same mutation was not found in another MH-susceptible individual whose CHCT was positive to the test with caffeine but not to the test with halothane. None of the 5 MH normal individuals of the family, previously diagnosed by CHCT, carry this mutation, nor do 100 controls from control Brazilian and USA populations. The Gly4932Ser variant is a candidate mutation for MH, based on its co-segregation with disease phenotype, absence among controls and its location within the protein.
Malignant hyperthermia; Mutation; Ryanodine receptor; Calcium channel
Braz J Med Biol Res, December 2009, Volume 42(12) 1218-1224
Multigenerational Brazilian family with malignant hyperthermia and a novel mutation in the RYR1 gene
A.R. Matos1, N. Sambuughin4, F.D. Rumjanek2, N.D. Amoedo2, L.B.P. Cunha3, G. Zapata-Sudo1 and  Correspondence and Footnotes
Correspondence and Footnotes
 R.T. Sudo1
R.T. Sudo1
1Programa de Desenvolvimento de Fármacos, Instituto de Ciências Biomédicas, 2Instituto de Bioquímica Médica, and 3Serviço de Anestesiologia, Faculdade de Medicina, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, Brasil
4Uniformed Services, University of the Health Sciences, Bethesda, MD, USA
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Malignant hyperthermia (MH) is a pharmacogenetic disease triggered in susceptible individuals by the administration of volatile halogenated anesthetics and/or succinylcholine, leading to the development of a hypermetabolic crisis, which is caused by abnormal release of Ca2+ from the sarcoplasmic reticulum, through the Ca2+ release channel ryanodine receptor 1 (RyR1). Mutations in the RYR1 gene are associated with MH in the majority of susceptible families. Genetic screening of a 5-generation Brazilian family with a history of MH-related deaths and a previous MH diagnosis by the caffeine halothane contracture test (CHCT) in some individuals was performed using restriction and sequencing analysis. A novel missense mutation, Gly4935Ser, was found in an important functional and conserved locus of this gene, the transmembrane region of RyR1. In this family, 2 MH-susceptible individuals previously diagnosed with CHCT carry this novel mutation and another 24 not previously diagnosed members also carry it. However, this same mutation was not found in another MH-susceptible individual whose CHCT was positive to the test with caffeine but not to the test with halothane. None of the 5 MH normal individuals of the family, previously diagnosed by CHCT, carry this mutation, nor do 100 controls from control Brazilian and USA populations. The Gly4935Ser variant is a candidate mutation for MH, based on its co-segregation with disease phenotype, absence among controls and its location within the protein.
Key words: Malignant hyperthermia; Mutation; Ryanodine receptor; Calcium channel
Introduction
Malignant hyperthermia (MH) is an autosomal dominant pharmacogenetic disease (1,2). Individuals who are susceptible to MH develop a hypermetabolic crisis after the administration of volatile general anesthetics and/or succinylcholine, with signs of tachycardia, arrhythmia, muscle rigidity, and hyperthermia. This life-threatening condition is caused by the increased release of Ca2+ from the sarcoplasmic reticulum to the cytoplasm by the ryanodine receptor 1 (RyR1) (3).
MH susceptibility has been linked to several loci in the genome (4-8), but most cases are associated with the RYR1 gene located on chromosome 19 (9,10), one of the largest and most complex genes in the human genome, comprising 106 exons and transcribing a 15,117 nucleotide-long RNA (11), which encodes a 563-kD homotetrameric protein. Worldwide, about 70-80% of MH-susceptible (MHS) individuals have mutations in RYR1 (12-15).
More than 180 different mutations (16) and many polymorphisms (14,15) have been described in RYR1 among MHS families. These mutations are frequently clustered in the hot-spot domains of the protein, which are the N-terminal region (amino acids 34-614), the central region (amino acids 2163-2458) and the C-terminal region (amino acids 4136-4973) (16).
Currently, MH is diagnosed by the contracture test, which is the diagnostic model adopted at the Brazilian MH Center. The caffeine-halothane contracture test (CHCT) is applied according to the North American protocol (17). However, because of its invasiveness, this test presents some disadvantages and, especially in Brazil, which has only one center for MH diagnosis, patients frequently also need to travel long distances and to spend much time and effort in order to undergo this test. All of these circumstances create great difficulties in the access to the diagnosis of MH in Brazil.
Molecular tests based on genetic analysis are desirable because they are less invasive and more informative for patients, especially when familial mutation is known. However, the complexity of this gene and the increasing number of reported mutations limit routine screening of RYR1 in MHS families. To this end, the development of a genetic test is underway in several countries, as well as the elaboration of guidelines applicable to molecular genetics (18,19).
A previous study of a Brazilian MH family revealed a new mutation in RYR1, Arg2355Cys (20), which permitted a genetic diagnosis according to the guidelines proposed in other members of the family that could not undergo CHCT. Genetic screening for RYR1 was reported thereafter in other Brazilian families (21,22). The Brazilian MH Center has registered many other families who are affected by this disorder but have not been investigated genetically.
In the present investigation, we studied a large multi- generational Brazilian family, in which several deaths and cases of hypermetabolic crisis under general anesthesia had occurred and were associated with MH. First, an MHS member of the family diagnosed by CHCT was screened for mutations in the RYR1 gene. Then, the genetic diagnosis test was applied to the rest of the family.
Subjects and Methods
Samples
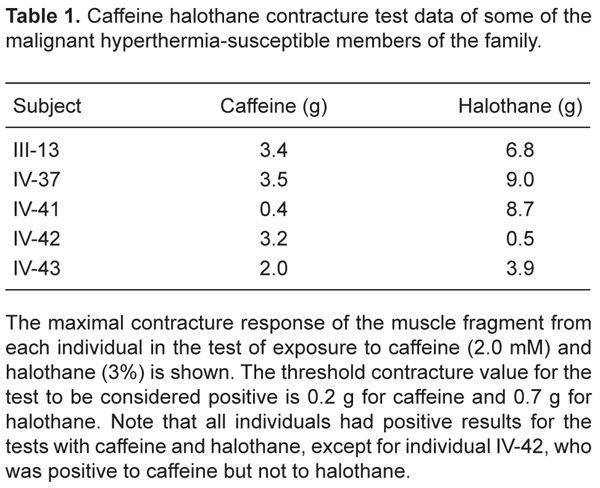
Blood samples from 101 individuals of a Brazilian family were sent to the Brazilian MH Center of Universidade Federal do Rio de Janeiro (UFRJ) for analysis. Eight of these individuals had already been diagnosed by CHCT according to the North American protocol (17) at the Brazilian MH Center of UFRJ. Three of them (IV-37, IV-41 and IV-42) were diagnosed as MHS and 5 (III-5, III-7, III-11, IV-30, and IV-31) were diagnosed as MH normal. It should be noted that IV-42 had a positive result for the test with caffeine but a negative result for the test with halothane, being diagnosed as MHS. The clinical history of the family describes death that could be associated with MH in II-1 and III-1 and with a hypermetabolic crisis after general anesthesia in IV-54.
Mutation screening
Genomic DNA was extracted from whole blood samples using the phenol-chloroform method (23). All the exons within the three mutational hot spots were sequenced using the Big DyeTerminator™ Cycle Sequencing kit and ABI 3100 Genetic Analyzer (Perkin Elmer, USA). A novel variant was found in exon 102 of RYR1. Primers 5'-cctcc ctcagtgttacctgttt-3' and 5'-GCCCAAGGTCTCAC AGTAGC-3' were used for PCR amplification of a 296-bp fragment that includes the locus of the mutation in exon 102. In some members of the family investigated, this mutation was also detected by restriction analysis. DNA sequence analysis revealed that the mutation created the restriction site GGATC for the enzyme HinfI. DNA fragments were then digested with HinfI (Invitrogen, USA) according to the manufacturer protocol, at 37°C for 1 h. Restriction fragments were then fractionated by agarose gel electrophoresis and visualized by staining with ethidium bromide (GE Health Sciences, USA). Next, the fragments of exon 102 from 19 members of the family chosen at random were sequenced in both directions at Macrogen Inc. (Korea) to confirm the results of restriction analysis. DNA sequencing was analyzed using the Contig and Align from Vector program (available at www.invitrogen.com). In addition, 100 control subjects from Brazilian and USA populations were screened for this variant.
Results
CHCT data for the MHS individuals of the family are presented in Table 1, including individual IV-42, who had a positive contracture response to the test with caffeine (3.2 g - threshold: 0.2 g), but not for the test with halothane (0.5 g - threshold: 0.7 g). The clinical data of the family revealed an MH reaction under general anesthesia for individual IV-54 and an unexplained sudden death associated with MH for individuals II-1 and III-1. The complete pedigree for six generations of this family is shown in Figure 1.
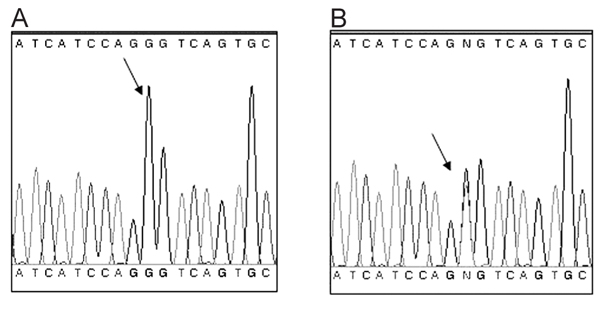
After screening for the hot spots of the RYR1 gene, a novel variant was found in exon 102, producing the missense mutation Gly4935Ser, which corresponds to a G14803A change at the DNA level. Furthermore, this alteration creates a new restriction site for the enzyme HinfI, which was used to investigate all the members of the family. After restriction analysis of the DNA fragment containing exon 102, the occurrence of the missense mutation was detected by the presence of additional fragments of 166 and 130 bp, and the original fragment of 296 bp, visualized by agarose gel electrophoresis (Figure 2). To confirm these results, 19 samples chosen at random were sequenced in both directions as described. This mutation was found in 26 members of this family. The electropherograms from individuals displaying or not the mutation are shown in Figure 3.
This mutation was found in individuals IV-37 and IV-41, who had been previously diagnosed as MHS by CHCT, but not in individual IV-42, who was confirmed by experiments of DNA restriction and sequencing analysis, also diagnosed as MHS by CHCT. The results in Table 1 show that the CHCT results for individuals IV-37 and IV-41 were positive for the test with caffeine and halothane, while the results for individual IV-42 were only positive for the test with caffeine. Individuals III-13 and IV-43 DNAs were not available.
One hundred and one individuals from the family were screened for the Gly4935Ser variant, which was found in 26, including 2 MHS individuals previously diagnosed by CHCT. This variant was not found in any of the 5 individuals who had been previously diagnosed by CHCT as MH normal (III-5, III-7, III-11, IV-30, and IV-31). Screening for this variant in 100 control subjects, including Brazilian and USA populations, showed negative results.
Pedigree of the Brazilian family of this study. The green symbols indicate individuals diagnosed as malignant hyperthermia (MH)-susceptible by the caffeine halothane contracture test (CHCT). The light blue symbols indicate individuals diagnosed as MH normal by CHCT. The red symbols indicate individuals who carry the Gly4935Ser mutation. The dark blue symbols indicate individuals who do not carry this mutation. The open symbols indicate individuals who were not studied by either the DNA test or CHCT. Note that 6 children descending from the first individual (I-1) generated the rest of the pedigree (II-1 to II-6).
Photographs of 1.2% agarose gels stained with ethidium bromide and visualized under UV light. A, PCR amplification; note that the arrow shows the 296-bp PCR fragment resulting from the individuals indicated above the panel. B, Restriction analysis of the DNA fragments with the enzyme HinfI, which cuts the fragments that carry the mutation. Note that the DNA fragments from individuals IV-46, IV-47, IV-51, IV-52, and IV-39 do not have their resulting band cut by the enzyme because no additional band was observed, indicating absence of the mutation. Individuals IV-53, IV-54, IV-55, IV-56, and IV-38 had their resulting band cut by Hinf because they all have two additional bands (166 and 130 bp) and the original band fragment (296 bp), indicating the presence of the mutation in one allele of the gene. 100 bp - Molecular weight marker.
DNA sequencing of the fragment generated by PCR, which contains a region of exon 102 of the RYR1 gene including the locus of the mutation. The Gly4935Ser mutation corresponds to a G14803A change at the DNA level. A, Electropherogram from an individual without this RYR1 mutation. The arrow shows a single peak corresponding to a G at nucleotide position 14803. B, Electropherogram from an individual carrying the RYR1 mutation. In contrast, there are two superposed peaks, which correspond to a G and an A at nucleotide position 14803, where the program shows the letter N (arrow). This indicates that this person is heterozygous for this mutation.
Comparison of the amino acid sequences of the region of the mutation in RyR1 found in the family. Note that this is a conserved domain among different species and isoforms of RyR, as well as in the IP3R protein. Letters in bold and underlined correspond to the site of the mutation. Normal letters are the conserved amino acids. Letters in italics correspond to non-conserved amino acids in the sequence.
Caffeine halothane contracture test data of some of the malignant hyperthermia-susceptible members of the family.
Discussion
We studied a large Brazilian family distributed along six generations with a history of MH. Some of the members were suspected to have died due to MH and others were diagnosed as MHS by CHCT. A novel amino acid variant, Gly4935Ser, was identified by direct sequencing. This variant was found in 26 members of the family, including two previously diagnosed MHS, individuals IV-37 and IV-41. The CHCT of these individuals were both positive to halothane and caffeine (Table 1), showing agreement of the phenotype with the genotype results.
In the other MHS individual diagnosed, IV-42, this mutation was not found, as confirmed by experiments of DNA restriction and sequencing analysis. This individual was sensitive to caffeine but not to halothane during the CHCT, a fact that raised the possibility of a false-positive test for this subject. According to the North American protocol, a person is diagnosed as MHS if he shows a positive result to one of the substances of the test (17). However, according to the European MH Group (EMHG) protocol, a person is diagnosed as equivocal for MH when only one of the tests shows a positive result (24).
It should be considered that CHCT has a high sensitivity of 99%, but a lower specificity of 78% for the North American protocol (25). Discordance between CHCT and genetic studies has already been reported (26-30), indicating that some familial mutations are not found in some MHS members of the family. It is also possible that this discordant individual has another mutation in another part of RYR1 not analyzed or in another gene related to MH (4-7). Some families that display more than one causative mutation leading to MH have been reported (16,31).
The missense mutation Gly4935Ser is located in exon 102, inside the last hot spot of the gene. This region has been reported to be less frequently mutated than the others (32). The position Gly4935 is evolutionarily conserved and also conserved between the other two isoforms of RyR and the IP3R protein (Figure 4) and its mutation was absent among the 100 control subjects studied here.
The C-terminus of the protein is a hydrophobic region that contains the conduction pore of the Ca2+ release channel (33), the transmembrane segments (34,35), one of the binding sites for Ca2+ (36), and the binding site for ryanodine (37). According to a proposed model (38), the position of the Gly4935 is at the end of the last transmembrane domain of RyR1, very close to the C-terminus of the protein.
We conclude that the Gly4935Ser variant is a candidate mutation for MHS in this Brazilian family, based on its co-segregation with disease phenotype, absence among controls and its location within the protein.
The CHCT is not easily available in Brazil. Its limited availability and invasiveness prevented its use in high-risk family members. Our study is an example of the possible use of a molecular genetic test for the diagnosis and prevention of MH. A genetic test for MH is especially important for large families in which many members, particularly children, can benefit without undergoing an invasive contracture test. This is even more important because, as a result of the speed and ease with which the analysis is carried out, a more comprehensive profile can be achieved.
Acknowledgments
This study was supported in part by FAPERJ, CNPq, CAPES, Cristália Produtos Químicos e Farmacêuticos Ltda., FUJB, and by CNPq fellowships (A.R. Matos, G. Zapata-Sudo, R.T. Sudo).
Address for correspondence: R.T. Sudo, Programa de Desenvolvimento de Fármacos, Instituto de Ciências Biomédicas, CCS, UFRJ, Bloco J, sala 14, 21941-590 Rio de Janeiro, RJ, Brasil. Fax: +55-21-2562-6505. E-mail: rtsudo@farmaco.ufrj.br
Received August 14, 2009. Accepted October 28, 2009. Available online November 13, 2009. Published December 4, 2009.
The Brazilian Journal of Medical and Biological Research is partially financed by






- 1. Britt BA, Locher WG, Kalow W. Hereditary aspects of malignant hyperthermia. Can Anaesth Soc J 1969; 16: 89-98.
- 2. Kalow W. Inheritance of malignant hyperthermia: a review of published data. In: Britt BA (Editor), Malignant hyperthermia Amsterdam: Martinus-Nijhoff; 1987. p 155-179.
- 3. Mickelson JR, Louis CF. Malignant hyperthermia: excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol Rev 1996; 76: 537-592.
- 4. Levitt RC, Olckers A, Meyers S, Fletcher JE, Rosenberg H, Isaacs H, et al. Evidence for the localization of a malignant hyperthermia susceptibility locus (MHS2) to human chromosome 17q. Genomics 1992; 14: 562-566.
- 5. Iles DE, Lehmann-Horn F, Scherer SW, Tsui LC, Olde WD, Suijkerbuijk RF, et al. Localization of the gene encoding the alpha 2/delta-subunits of the L-type voltage-dependent calcium channel to chromosome 7q and analysis of the segregation of flanking markers in malignant hyperthermia susceptible families. Hum Mol Genet 1994; 3: 969-975.
- 6. Sudbrak R, Procaccio V, Klausnitzer M, Curran JL, Monsieurs K, van BroeckhovenC, et al. Mapping of a further malignant hyperthermia susceptibility locus to chromosome 3q13.1. Am J Hum Genet 1995; 56: 684-691.
- 7. Gregg RG, Couch F, Hogan K, Powers PA. Assignment of the human gene for the alpha 1 subunit of the skeletal muscle DHP-sensitive Ca2+ channel (CACNL1A3) to chromosome 1q31-q32. Genomics 1993; 15: 107-112.
- 8. Robinson RL, Monnier N, Wolz W, Jung M, Reis A, Nuernberg G, et al. A genome wide search for susceptibility loci in three European malignant hyperthermia pedigrees. Hum Mol Genet 1997; 6: 953-961.
- 9. Jurkat-Rott K, McCarthy T, Lehmann-Horn F. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve 2000; 23: 4-17.
- 10. McCarthy TV, Quane KA, Lynch PJ. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat 2000; 15: 410-417.
- 11. Phillips MS, Fujii J, Khanna VK, DeLeon S, Yokobata K, de Jong PJ, et al. The structural organization of the human skeletal muscle ryanodine receptor (RYR1) gene. Genomics 1996; 34: 24-41.
- 12. Sambuughin N, Holley H, Muldoon S, Brandom BW, de Bantel AM, Tobin JR, et al. Screening of the entire ryanodine receptor type 1 coding region for sequence variants associated with malignant hyperthermia susceptibility in the North American population. Anesthesiology 2005; 102: 515-521.
- 13. Ibarra MC, Wu S, Murayama K, Minami N, Ichihara Y, Kikuchi H, et al. Malignant hyperthermia in Japan: mutation screening of the entire ryanodine receptor type 1 gene coding region by direct sequencing. Anesthesiology 2006; 104: 1146-1154.
- 14. Galli L, Orrico A, Lorenzini S, Censini S, Falciani M, Covacci A, et al. Frequency and localization of mutations in the 106 exons of the RYR1 gene in 50 individuals with malignant hyperthermia. Hum Mutat 2006; 27: 830.
- 15. Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 2006; 27: 977-989.
- 16. Nelson TE. Malignant hyperthermia: a pharmacogenetic disease of Ca++ regulating proteins. Curr Mol Med 2002; 2: 347-369.
- 17. Larach MG. Standardization of the caffeine halothane muscle contracture test. North American Malignant Hyperthermia Group. Anesth Analg 1989; 69: 511-515.
- 18. Urwyler A, Deufel T, McCarthy T, West S. Guidelines for molecular genetic detection of susceptibility to malignant hyperthermia. Br J Anaesth 2001; 86: 283-287.
- 19. Nelson TE, Rosenberg H, Muldoon SM. Genetic testing for malignant hyperthermia in North America. Anesthesiology 2004; 100: 212-214.
- 20. McWilliams S, Nelson T, Sudo RT, Zapata-Sudo G, Batti M, Sambuughin N. Novel skeletal muscle ryanodine receptor mutation in a large Brazilian family with malignant hyperthermia. Clin Genet 2002; 62: 80-83.
- 21. Muniz VP, Silva HC, Tsanaclis AM, Vainzof M. Screening for mutations in the RYR1 gene in families with malignant hyperthermia. J Mol Neurosci 2003; 21: 35-42.
- 22. Kossugue PM, Paim JF, Navarro MM, Silva HC, Pavanello RC, Gurgel-Giannetti J, et al. Central core disease due to recessive mutations in RYR1 gene: is it more common than described? Muscle Nerve 2007; 35: 670-674.
- 23. Sambrook J, Russell DW. Rapid isolation of yeast DNA. In: Anonymous, Molecular cloning, a laboratory manual New York: Cold Spring Harbor Laboratory; 2001. p 631-632.
- 24. A protocol for the investigation of malignant hyperpyrexia (MH) susceptibility. The European Malignant Hyperpyrexia Group. Br J Anaesth 1984; 56: 1267-1269.
- 25. Allen GC, Larach MG, Kunselman AR. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. The North American Malignant Hyperthermia Registry of MHAUS. Anesthesiology 1998; 88: 579-588.
- 26. Deufel T, Sudbrak R, Feist Y, Rubsam B, Du Chesne I, Schafer KL, et al. Discordance, in a malignant hyperthermia pedigree, between in vitro contracture-test phenotypes and haplotypes for the MHS1 region on chromosome 19q12-13.2, comprising the C1840T transition in the RYR1 gene. Am J Hum Genet 1995; 56: 1334-1342.
- 27. Serfas KD, Bose D, Patel L, Wrogemann K, Phillips MS, MacLennan DH, et al. Comparison of the segregation of the RYR1 C1840T mutation with segregation of the caffeine/halothane contracture test results for malignant hyperthermia susceptibility in a large Manitoba Mennonite family. Anesthesiology 1996; 84: 322-329.
- 28. Robinson RL, Anetseder MJ, Brancadoro V, van Broekhoven C, Carsana A, Censier K, et al. Recent advances in the diagnosis of malignant hyperthermia susceptibility: how confident can we be of genetic testing? Eur J Hum Genet 2003; 11: 342-348.
- 29. Brown RL, Pollock AN, Couchman KG, Hodges M, Hutchinson DO, Waaka R, et al. A novel ryanodine receptor mutation and genotype-phenotype correlation in a large malignant hyperthermia New Zealand Maori pedigree. Hum Mol Genet 2000; 9: 1515-1524.
- 30. Monnier N, Kozak-Ribbens G, Krivosic-Horber R, Nivoche Y, Qi D, Kraev N, et al. Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Hum Mutat 2005; 26: 413-425.
- 31. Monnier N, Krivosic-Horber R, Payen JF, Kozak-Ribbens G, Nivoche Y, Adnet P, et al. Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology 2002; 97: 1067-1074.
- 32. Sei Y, Sambuughin NN, Davis EJ, Sachs D, Cuenca PB, Brandom BW, et al. Malignant hyperthermia in North America: genetic screening of the three hot spots in the type I ryanodine receptor gene. Anesthesiology 2004; 101: 824-830.
- 33. Bhat MB, Zhao J, Takeshima H, Ma J. Functional calcium release channel formed by the carboxyl-terminal portion of ryanodine receptor. Biophys J 1997; 73: 1329-1336.
- 34. Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature 1989; 339: 439-445.
- 35. Zorzato F, Fujii J, Otsu K, Phillips M, Green NM, Lai FA, et al. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem 1990; 265: 2244-2256.
- 36. Callaway C, Seryshev A, Wang JP, Slavik KJ, Needleman DH, Cantu C III, et al. Localization of the high and low affinity [3H]ryanodine binding sites on the skeletal muscle Ca2+ release channel. J Biol Chem 1994; 269: 15876-15884.
- 37. Witcher DR, McPherson PS, Kahl SD, Lewis T, Bentley P, Mullinnix MJ, et al. Photoaffinity labeling of the ryanodine receptor/Ca2+ release channel with an azido derivative of ryanodine. J Biol Chem 1994; 269: 13076-13079.
- 38. Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH. Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1). Proc Natl Acad Sci U S A 2002; 99: 16725-16730.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
04 Dec 2009 -
Date of issue
Dec 2009
History
-
Accepted
28 Oct 2009 -
Received
14 Aug 2009