Abstract
Micro-ribonucleic acids (microRNAs) are small molecules containing 20-23 nucleotides. Despite their small size, it is likely that almost every cellular process is regulated by them. Moreover, aberrant microRNA expression has been involved in the development of various diseases, including cancer. Although many data are available about the role of microRNAs in various lymphoproliferative disorders, their impact on the development of acute lymphoblastic leukemia of T-cell progenitors is largely unknown. In this review, we present recent information about how specific microRNAs are expressed and regulated during malignant T-lymphopoiesis and about their role during normal hematopoiesis.
MicroRNAs; Gene expression; Leukemia; Acute lymphoblastic leukemia; T cells
Braz J Med Biol Res, July 2010, Volume 43(7) 619-626
The role of micro-ribonucleic acids in normal hematopoiesis and leukemic T-lymphogenesis
S.N. Slavov, H.L. Gimenes Teixeira and
Correspondence and Footnotes
 E.M. Rego
E.M. Rego
Divisão de Hematologia/Oncologia, Departamento de Clínica Médica, Faculdade de Medicina de Ribeirão Preto e Instituto Nacional de Ciência e Tecnologia em Células-Tronco e Terapia Celular, Universidade de São Paulo, Ribeirão Preto, SP, Brasil
 Text
Text
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Micro-ribonucleic acids (microRNAs) are small molecules containing 20-23 nucleotides. Despite their small size, it is likely that almost every cellular process is regulated by them. Moreover, aberrant microRNA expression has been involved in the development of various diseases, including cancer. Although many data are available about the role of microRNAs in various lymphoproliferative disorders, their impact on the development of acute lymphoblastic leukemia of T-cell progenitors is largely unknown. In this review, we present recent information about how specific microRNAs are expressed and regulated during malignant T-lymphopoiesis and about their role during normal hematopoiesis.
Key words: microRNAs; Gene expression; Leukemia; Acute lymphoblastic leukemia; T cells
Text

Introduction
For more than 50 years the term "gene" has been synonymous of genomic regions encoding messenger RNAs (mRNAs). However, recent studies have demonstrated that the entire genome gives rise not only to mRNAs but also to thousands of small regulatory RNAs, the so-called non-coding micro RNAs (microRNAs) (1). The first microRNA was described by the group of V. Ambros in the nematode Caenorhabditis elegans and was named lin-4 (2). Five years later, in 1998, the process of RNA interference (silencing of specific genes) carried out by microRNAs was described in plants (3,4). Subsequently, it was assumed that microRNAs regulate gene expression at the post-transcriptional level in presumably every multicellular organism (5). At present, it is clear that more than 3% of human genes encode microRNAs, regulating approximately 90% of the protein-coding genes (6).
microRNAs consist of small molecules containing 20-23 nucleotides (nt) and function most frequently as negative regulators of gene expression. They exert their effects post-transcriptionally by inhibiting mRNA translation or inducing mRNA degradation, and thus participate in a wide variety of physiological and pathological cellular functions (7). microRNAs are now among the most studied non-coding RNAs and it is not an exaggeration to say that their identification opened a new era in cancer research. Over the past few years, molecular oncology research has revealed that abnormalities in both protein-coding genes and microRNAs can be identified in tumors and that the interplay between proteins and microRNAs is involved in the initiation, progression, and metastasis of various types of human cancer (8). Such malignancies include brain, liver, prostate, ovarian, and oral cancer, as well as lymphomas, but this list is probably much longer (9-14). Aberrant microRNA expression could also play a vital role in the pathogenesis of leukemia and thus microRNAs have rapidly emerged as potential targets for anti-leukemia therapy (10).
This review focuses on recent information about the important roles of microRNAs during the normal hematopoietic development as well as their implications during the progress of leukemia and more specifically of T-cell malignancies.
Biogenesis and cellular functions of microRNAs
microRNAs are encoded within different genomic regions including protein-coding genes and non-coding transcription units. Approximately 50% of all synthesized microRNAs are derived from non-coding RNA transcripts, while an additional ~40% are located at introns of the protein-coding genes. The majority of microRNAs are transcribed by RNA polymerase II (RNA pol II) and bear a 7-methyl guanidine cap at the 5’-end of the molecule and a poly-adenine (poly-A) tail at its 3’ end (15).
The mechanism giving rise to mature microRNAs involves two processing cleavages by the RNAse III enzymes Drosha and Dicer. Following transcription by RNA pol II, Drosha processes the primary microRNA transcript (pri-microRNA) into ~60-100-nt hairpin structure designated as precursor RNA (pre-microRNA). Through interaction with exportin-5 and Ran-GTP, the pre-microRNA is transported into the cytoplasm, where it is processed de novo by the catalytic activity of Dicer. This ultimate cleavage gives rise to a double-stranded ~20-23-nt product, which consists of a mature microRNA guide strand and a passenger strand (microRNA*). The mature microRNA is then loaded into effector complexes known as RNA-induced silencing complex, while the passenger strand is degraded (16).
microRNAs inhibit gene expression by binding to the complementary 3’ untranslated region (3’UTR) of the target messenger RNA. Every organism synthesizes hundreds of unique microRNAs, each predicted to regulate specific target genes. Moreover, nearly all microRNAs identified in humans, plants and animals are evolutionary conserved (17).
Functions of microRNA in normal hematopoiesis
Hematopoiesis is sustained by continuous differentiation of multipotent hematopoietic stem cells (HSCs) into hematopoietic progenitor cells (HPCs), giving rise to different lineages and mature blood cells. Regulation of hematopoiesis consists of two aspects: self-renewal of HSCs (progressive restriction of lineage potential) and differentiation of HSCs/HPCs (development into differentiated precursors up to mature cells). HSC differentiation leads to the formation of multipotent cells, which have largely lost their capacity for self-renewal, but retain the ability to differentiate into many types of blood cells. These HPCs generate common lymphoid progenitors, giving rise to B- and T-lymphocytes, and common myeloid progenitors, generating erythrocytes, megakaryocytes, granulocytes, and monocytes/macrophages (Figure 1) (18).
microRNAs are differentially expressed during normal hematopoiesis (Figure 1). Chen et al. (19) were the first to report that microRNAs are specifically expressed and dynamically regulated during murine hematopoietic cell development (19). Since that report, several groups have used different techniques to isolate progenitors, to induce uni- or multilineage differentiation, and to describe the microRNA expression profile and/or function during murine and human hematopoiesis.
During human erythropoiesis, microRNA-221 and microRNA-222 are highly expressed in CD34+ HPCs and are gradually down-modulated during unilineage erythroid differentiation. The decline of microRNA-221 and microRNA-222 expression during the exponential erythroid growth unblocks c-kit protein translation. Since c-kit is a receptor of the kit ligand or stem cell factor, this can lead to expansion of early erythroblasts (20). Two additional pathways essential for erythropoiesis are also modulated by microRNAs: ALK4 and GATA-1. MicroRNA-24 is a negative regulator that inhibits erythroid differentiation by targeting active type I receptor ALK4 (21). The second pathway is a globin transcription factor 1 (GATA-1-regulated microRNA locus, consisting of microRNA-144 and microRNA-451, which regulates erythroid development in zebrafish embryos (22).
With respect to the common erythrocyte-megakaryocyte precursors, microRNA-150 was found to drive in vitro and in vivo megakaryocytic differentiation at the expense of the erythroid lineage (23). Furthermore, a regulatory pathway in megakaryocytopoiesis involving the promyelocytic leukemia zinc finger (PLZF) transcription factor, microRNA-146a and the stromal-cell-derived factor 1 receptor CXCR4 has been identified. microRNA-146a was initially reported to be an endotoxin-responsive gene in human monocytes. PLZF protein is a transcription factor that is involved in the regulation of limb and skeleton formation, hematopoietic proliferation and differentiation, leukemogenesis, and tumorigenesis. In this recently identified pathway, PLZF transcriptionally inhibits microRNA-146a, which in turn regulates the CXCR4 co-receptor. In CD34+ HPC megakaryopoietic cultures, PLZF was up-regulated, a fact leading to a decrease of microRNA-146a expression and to increased levels of CXCR4 protein. MicroRNA-146a overexpression as well as PLZF or CXCR4 silencing impaired megakaryocyte proliferation, differentiation, maturation, and colony formation (24-26).
The key role of microRNAs was also demonstrated in human monocytopoiesis by two separate studies (27). The first examined the role of microRNAs of the 17-5p-92 and 106a-92 clusters in monocytic differentiation (28). The microRNA-17-5p-92 cluster consists of seven microRNAs located on chromosome 13 and transcribed as a polycistronic unit (15). This cluster is highly homologous to the microRNA-106a-92 and microRNA-106b-25 clusters located on chromosomes Y and 7, respectively (29). During monocytic differentiation, low levels of microRNA-17-5p/20a/106a allow translation of acute myeloid leukemia 1 (AML1) transcription factor that inhibits microRNA-17-5p-92 and microRNA-106a-92 clusters. It also trans-activates the expression of monocyte colony-stimulating factor (M-CSF), thus inducing monocytic cell differentiation. This study indicates that monocytopoiesis is controlled by a circuitry sequentially involving microRNA-17-5p/20a/106a, AML1 and M-CSF receptor, whereby microRNA-17-5p/20a/106a functions as a gene complex interlinked with AML1 in a mutual negative-feedback loop (27). Interestingly, the microRNA-17-92 cluster is also essential for B-lymphocyte development in vivo. Its targeted deletion in mice resulted in lung hypoplasia and inhibition of the pro-B to pre-B transition (30). On the other hand, transgenic expression of microRNA-17-92 in mice caused the development of lymphoproliferative diseases and autoimmunity disorders (31).
Additional microRNAs are also involved in the PU.1-dependent regulatory pathway, which is required for normal human monocytic differentiation (28). It was found that the master transcription factor PU.1 directly activates microRNA-424 transcription during monocytic differentiation of promyelocytic blasts as well as human HPCs in unilineage monocyte cultures. microRNA-424 was found to be a potent inducer of monocyte differentiation, particularly by targeting the transcription nuclear factor I-A (NFI-A), whose down-regulation is critical for the progression to monocytic differentiation (28).
microRNAs have also been linked to macrophage functions during inflammation and development of innate immunity. microRNA-155 was found to be up-regulated in primary murine macrophages after exposure to interferon β and poly(I:C) (32). Sustained expression of microRNA-155 in HSCs causes abnormal proliferation of myeloid cells (33).
During myelopoiesis, microRNA-223 levels rise progressively and their suppression blocks granulocytic maturation. The levels of microRNA-233 are modulated by competitive binding to its upstream part of two CCAAT-box-binding proteins: NFI-A and C/EPBα. These two proteins bind to the CAAT elements located on the pre-microRNA promoter. NFI-A maintains microRNA-223 at low levels, while its replacement by C/EPBα activation results in microRNA-223 up-regulation and granulocytic differentiation (34). The transcriptional induction of the myeloid-specific microRNA-223 is responsible for granulocyte-monocyte precursor progenitor proliferation and granulocyte function in vivo (35,36).
microRNA expression and significance during normal and malignant T-lymphopoiesis
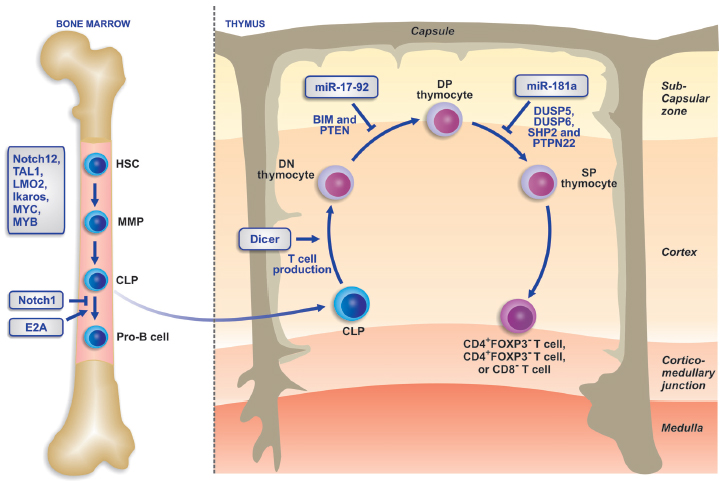
The earliest progenitors of T-lymphocytes are derived from the bone marrow. They are extremely rare and typically are present within complex populations of hematopoietic stem cells (37). Although they are generated within the bone marrow, most of T-cell development occurs in a specialized organ, i.e., the thymus. These distinct differentiation compartments underscore the unique capacity of the thymic microenvironment to support T-cell lineage restriction and differentiation. The development of T-lymphocytes is supported by many thymus-derived signals such as Notch, morphogenetic, and protein kinase signals (38). The development of T cells in the thymus as well as their activation at the periphery, are controlled by complex protein signaling pathways that are regulated by the microRNAs. Expression profiles of these microRNAs vary during the different stages of T cell and cellular subset development. Two specific microRNAs, microRNA-17-92 and microRNA-155, have been implicated in T-cell development and probably account for some of the phenotype of Dicer deficiency in T cells. Recent data also indicate a role of microRNAs in the differentiation of T cells into different effector T-helper cell subsets. For example, in microRNA-155-deficient mice, T cells develop directly into Th2 cells, which indicate that microRNA-155 regulates the differentiation into Th type 1 cells. Also, mice with conditional deletion of Dicer or Drosha in regulatory T cells (Treg) show requirement for the microRNA pathway in forkhead boxP3+ Treg cells. These animals develop a lethal autoimmune inflammatory disease accompanied by impaired development and function of Treg cells (39). Moreover, mice lacking microRNA-155 were immunodeficient and displayed increased lung airway remodeling due to the impaired functions of B- and T-lymphocytes and dendritic cells (40). The development of T cells and the regulatory role of microRNAs is described in Figure 2 (41).
Recent data suggest that microRNA genes are new epigenetic targets in cancer development. Their altered regulation by oncogenic proteins and more specifically by fusion proteins expressed during hematological malignancies is potentially relevant to the pathogenesis of leukemic diseases. Numerous oncogenes and tumor suppressor genes are potentially regulated by microRNAs (42). microRNAs may play a role in oncogenesis as oncogenes, probably by variety of mechanisms involving the elimination of tumor suppressor proteins or suppression of genes by targeting oncogenic mRNAs (43).
microRNA-mediated tumorigenesis may result from either down-regulation of tumor suppressor genes or up-regulation of oncogenes. Usually microRNA genes are located at fragile sites of the genome as well as in minimal regions with loss of heterozygosity, regions of amplification (minimal amplicons) or common breakpoint regions (42). The aberrant expression at such sites has been associated with solid tumors and hematopoietic malignancies, as suggested by the following examples: frequent deletion of microRNA-15a and microRNA-16-1 in chronic lymphocytic leukemia, increased levels of microRNA-155 in diffuse large B-cell lymphomas, and increased levels of microRNA-181 in acute myeloid leukemia (AML) types M1 and M2 (43). Besides, aberrancies of the normal microRNome have been well documented in almost all hematological malignancies, suggesting in most cases the so-called specific microRNA-signatures. Also of importance is the fact that some of the microRNA-expression abnormalities described in leukemias and lymphomas can be interpreted on the basis of the microRNome variations during normal hematologic ontogenesis. This suggests that their origin is from a particular differentiation stage in which the hematopoietic differentiation is "frozen" in the malignancy (44).
Acute lymphoblastic leukemia (ALL) is a genetically heterogeneous lymphoid malignancy, which may involve B or T lineages, and leukemic cells may present immunophenotypic markers characteristic of distinct stages of maturation (45-47). The quantitative and qualitative differences in the expression of these markers are useful for the diagnosis, classification and prognostic stratification of patients with ALL (47). Moreover, there are geographic differences in the distribution of ALL subtypes (48,49). In addition to immunophenotypic markers, ALL is characterized by various underlying genetic abnormalities. Zanette et al. (50) have compared the profile of microRNA expression in CD19+ cells from patients with ALL of B-progenitors with CD19+ cells from the peripheral blood of healthy subjects. The five most highly expressed microRNAs in ALL were microRNA-128b, microRNA-204, microRNA-218, microRNA-331, and microRNA-181b-1. The most represented microRNA in ALL is microRNA-128b with more than a 400-fold difference compared to normal CD19+ B cells. microRNA-128b is a reported homologue of microRNA-128a and has been found in colon, lung, and pancreas solid cancers. On the other hand, the four microRNAs with the lowest expression levels were microRNA-135b, microRNA-132, microRNA-199s, microRNA-139, and microRNA-150 (50).
More recently, Mi et al. (51) performed a large-scale genome-wide microRNA expression profiling assay and identified 27 microRNAs that are differentially expressed between ALL and AML. Using expression signatures of at least two of these microRNAs results in an accuracy rate of >95% in distinguishing and diagnosing AML and ALL. Among these microRNAs differentially expressed between ALL and AML, microRNA-128a, microRNA-128b, let-7b, and microRNA-233 were most significantly distinct. microRNAs 128a and 128b were expressed at higher levels in ALL, whereas microRNAs 233 and let-7b were expressed predominantly in AML. microRNA-233 is also reported to be a "myeloid" gene that plays a critical role in myeloid functions and differentiation (51).
Interestingly, a special microRNA expression profile was recently identified in children experiencing ALL relapse, which was characterized by high levels of expression of microRNA-7, microRNA-198 and microRNA-663 and low levels of microRNA-126, microRNA-222, microRNA-551a, microRNA-345. Nevertheless, the mechanisms involved in the regulation of these microRNAs have not been identified. This "cascade" of microRNAs was also identified in samples of ALL with subsequent central nervous system relapse, suggesting they may serve as biomarkers in the detection of early central nervous system relapse of pediatric ALL. Interestingly, target prediction of the microRNA pattern revealed that some abnormally expressed microRNAs may putatively target neuron function- and neurotransmitter-related genes (52).
The prognostic value of microRNAs in ALL is further demonstrated by the significance of microRNA-16. In general, low levels of microRNA-16 expression are associated with better outcome, while higher expression levels of this microRNA are associated with the poorest type of prognosis. This microRNA functions by inhibiting cell cycle progression by targeting several cyclins (E, D1), CDK6 and by inducing apoptosis by inhibiting BCL-2 translation. Considering these main functions of microRNA-16, the poor prognosis associated with its higher expression levels in ALL is astonishing. In this respect, researchers have failed to demonstrate a relationship between microRNA-16 expression and malignant lymphocyte proliferation or differentiation. However, investigation of the extent to which microRNA-16 varies within normal peripheral blood leukocyte stimulation, has revealed that its decrease after stimulation is necessary for the induction of lymphocyte proliferation. In contrast, a microRNA-16 increase might act as a restriction point in the entire proliferation process. Thus, microRNA-16 expression levels appear to be of prognostic significance in ALL, which may be controlled in independent and larger series (53).
We have recently studied the microRNA expression profile in a subgroup of patients with T-ALL, associated with a poor prognosis: T-ALL expressing NK markers (CD56 or N-CAM) (54,55). By multiple comparisons, we determined the microRNA expression profile of normal CD3+/CD56- and CD3-/CD56+ cells and of T-ALL/CD56+ and T-ALL/CD56- leukemic samples. Hierarchical cluster analysis revealed that normal and leukemic samples were grouped into two distinct clusters. The expression of 95 of 157 (60.5%) microRNAs did not differ from that of endogenous RNAs. Most of the differentially expressed microRNAs were less expressed in leukemic cells than in normal cells. microRNA-29b was significantly more expressed in leukemic cells than in normal cells. By contrast, microRNA-223 was less expressed in these comparisons. Known targets include the TCL1 and MCL1 genes for the former and NFIA-1 for the latter. Interestingly, the relevance of microRNA-223 to hematopoietic differentiation has been demonstrated in myelopoiesis, where there is a direct correlation between the transcriptional and epigenetic regulation of microRNA-223 (34-36).
In our study (54), microRNA-152 was less expressed in leukemic blasts compared to normal cells; however, within the latter, microRNA-152 was more expressed in CD3-/CD56+ than in CD3+/CD56- cells. microRNA-181a and microRNA-181b were more expressed in T-ALL/CD56- cells than in CD3+/CD56- cells, whereas microRNA-374 was more expressed in T-ALL/CD56+ cells than in CD3+/CD56- and CD3-/CD56+ cells. Comparison between T-ALL/CD56+ and T-ALL/CD56- cells demonstrated a 270-fold increase in microRNA-221 expression in the former. Among the microRNA-221 targets are p27/kip1, an important cell cycle regulator, and the c-kit receptor from the tyrosine kinase family, which have constitutively augmented activity in other hematological malignancies.
Mi et al. (56) have demonstrated that microRNAs-17-92 could play an essential role in the development of mixed lineage leukemia (MLL)-rearranged acute leukemia. This cluster was highly expressed not only in MLL-associated AML but also in ALL. The amplification of 13q31 and up-regulation by MLL fusions possibly contribute to the overexpression of the microRNA-17-92 cluster in MLL-rearranged leukemia. Moreover, microRNA-17-92 significantly increase cell viability, while inhibiting the apoptosis of HeLa (human cervix carcinoma) and 293T (human embryonal kidney) lineage cells and enhance proliferation of mouse normal bone marrow progenitor cells, resulting in the transformation of these cells by microRNA-17-92 and in cooperation with MLL fusions. This suggests that overexpression of the microRNA-17-92 cluster in MLL-rearranged leukemias is attributable to both DNA copy number amplification and direct up-regulation by MLL fusions.
The control of microRNA expression in ALL is still poorly understood. Nonetheless, several experimental data indicate that epigenetic regulation is an important mechanism guiding the regulation of microRNA expression in ALL. Recently, Li et al. (57) observed that histone modifications are involved in microRNA deregulation in human ALL cells. Using NALM-6 cells and primary leukemic cells, this group demonstrated that the silencing of the microRNA-22 gene in all cells is associated with the accumulation of histone modifications in its promoter element, independent of DNA methylation. microRNA-22 is frequently down-regulated in human ALL cells and the enforced expression of the microRNA-22 gene can inhibit the in vivo growth of B-cell lymphomas in mice, indicating the anti-tumor effect of microRNA-22. Moreover, histone modifications have been implicated in various cancers and histone deacetylase activity inhibitors possess significant growth inhibitory activity on ALL cells. For further research activities it would be of interest to investigate the functional role of microRNA-22 in histone modification associated with tumor initiation, promotion and progression of leukemia cells (57).
The regulation of microRNA expression is documented in cells infected with human retroviruses. Human T-cell leukemia virus type-1 (HTLV-1) is a causative agent of adult T-cell leukemia, which is an aggressive CD4+ malignancy. Tomita et al. (58) discovered that the expression of selected microRNAs and especially microRNA-146a is altered in HTLV-1-infected T-cell lines compared to uninfected cells. MicroRNA-146a is one of the key molecules in the inflammatory response and its expression is induced by activation by the NF-κB signaling pathway. It is expressed in HTLV-1-infected T-cell lines and is induced directly by the virus-encoded transactivator protein tax that plays a central role in viral pathogenecity. Exactly how the tax-microRNA-146a relationship regulates cell growth is difficult to determine, but it probably has several targets including IRAK 1, TRAF-6, IL-8, CXCR4, and matrix metalloproteinase 9. Thus, identification of aberrant expression of specific microRNAs in HTLV-1-infected cells may reveal novel retroviral functions in the pathogenesis of latent HTLV-1 infection. In the future, microRNAs could be used in clinical studies to predict the development of HTLV-1 leukemogenesis. Inhibition of microRNA-146a by anti-microRNA inhibitors suppresses the proliferation of HTLV-1-infected T-cell lines but, on the other hand, uninfected cell lines proliferate rapidly. These data suggest that microRNA-146a might be a promising therapeutic target in adult T-cell leukemia (58).
In conclusion, microRNAs are key regulators of cellular differentiation and their aberrant expression has been associated with the characteristic block of T-cell differentiation in T-ALL. Moreover, differential synthesis of some microRNAs has proved to be a prognostic factor for the outcome of ALL and/or could be used for its diagnosis. As a matter of fact, analysis of microRNA expression may prove to be of greater clinical significance than mRNA profiling, in contrast to mRNA expression, for a small number of microRNAs might be sufficient to classify human leukemias and, unlike mRNAs, microRNAs remain intact in routinely collected formalin-fixed or paraffin-embedded clinical tissues. Nevertheless, this assumption still requires extensive independent studies to be conducted before microRNA analysis is introduced into routine clinical exams.
Role of microRNAs in normal hematopoiesis. The figure shows the microRNAs involved in, and their expression during the hematopoiesis and differentiation of MPP cells into different lineages. Red and green color indicates microRNAs that are overexpressed and underexpressed, respectively. MPP = multipotent progenitor cell; CLP = common lymphoid progenitor; DN = double negative; DP = double positive; Th1 = T-helper 1; Th2 = T-helper 2; CMP = common myeloid progenitor; GMP = granulocyte-monocyte precursor; GP = granulocyte precursor; G = granulocyte; MP = monocyte-macrophage precursor; Mo = monocyte; Ma = macrophage; EMP = erythrocyte-megakaryocyte precursor; EP = erythrocyte precursor; E = erythrocyte; MgP = megakaryocyte precursor; Pl = platelets.
Schematic diagram of T-cell development and regulation and the participation of microRNAs in its regulation. HSC = hematopoietic stem cell; MMP = multipotent progenitor cell; CLP = common lymphoid precursors; DN thymocyte = double-negative thymocyte; DP thymocyte = double-positive thymocyte; SP thymocyte = single-positive thymocyte. (Reproduced from Ref. 41, with permission from Macmillian Publishers Ltd.).
References
1. Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol 2010; 220: 126-139.
2. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993; 75: 843-854.
3. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell 2009; 136: 642-655.
4. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391: 806-811.
5. Mueller DW, Bosserhoff AK. Role of miRNAs in the progression of malignant melanoma. Br J Cancer 2009; 101: 551-556.
6. Shah PP, Hutchinson LE, Kakar SS. Emerging role of microRNAs in diagnosis and treatment of various diseases including ovarian cancer. J Ovarian Res 2009; 2: 11.
7. Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett 2009; 285: 116-126.
8. Spizzo R, Rushworth D, Guerrero M, Calin GA. RNA inhibition, microRNAs, and new therapeutic agents for cancer treatment. Clin Lymphoma Myeloma 2009; 9 (Suppl 3): S313-S318.
9. Corney DC, Nikitin AY. MicroRNA and ovarian cancer. Histol Histopathol 2008; 23: 1161-1169.
10. Croce CM. MicroRNAs and lymphomas. Ann Oncol 2008; 19 (Suppl 4): iv39-iv40.
11. Gomes CC, Gomez RS. MicroRNA and oral cancer: future perspectives. Oral Oncol 2008; 44: 910-914.
12. Nicoloso MS, Calin GA. MicroRNA involvement in brain tumors: from bench to bedside. Brain Pathol 2008; 18: 122-129.
13. Shi XB, Tepper CG, White RW. MicroRNAs and prostate cancer. J Cell Mol Med 2008; 12: 1456-1465.
14. Varnholt H. The role of microRNAs in primary liver cancer. Ann Hepatol 2008; 7: 104-113.
15. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23: 4051-4060.
16. Davis BN, Hata A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun Signal 2009; 7: 18.
17. Tsuchiya S, Okuno Y, Tsujimoto G. MicroRNA: biogenetic and functional mechanisms and involvements in cell differentiation and cancer. J Pharmacol Sci 2006; 101: 267-270.
18. Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007; 7: 105-117.
19. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004; 303: 83-86.
20. Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci U S A 2005; 102: 18081-18086.
21. Wang Q, Huang Z, Xue H, Jin C, Ju XL, Han JD, et al. MicroRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood 2008; 111: 588-595.
22. Dore LC, Amigo JD, dos Santos CO, Zhang Z, Gai X, Tobias JW, et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc Natl Acad Sci U S A 2008; 105: 3333-3338.
23. Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell 2008; 14: 843-853.
24. Kelly KF, Daniel JM. POZ for effect - POZ-ZF transcription factors in cancer and development. Trends Cell Biol 2006; 16: 578-587.
25. Labbaye C, Spinello I, Quaranta MT, Pelosi E, Pasquini L, Petrucci E, et al. A three-step pathway comprising PLZF/miR-146a/CXCR4 controls megakaryopoiesis. Nat Cell Biol 2008; 10: 788-801.
26. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006; 103: 12481-12486.
27. Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, et al. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 2007; 9: 775-787.
28. Rosa A, Ballarino M, Sorrentino A, Sthandier O, De Angelis FG, Marchioni M, et al. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc Natl Acad Sci U S A 2007; 104: 19849-19854.
29. Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol 2004; 339: 327-335.
30. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008; 132: 875-886.
31. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 2008; 9: 405-414.
32. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A 2007; 104: 1604-1609.
33. O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med 2008; 205: 585-594.
34. Nervi C, Fazi F, Grignani F. Oncoproteins, heterochromatin silencing and microRNAs: a new link for leukemogenesis. Epigenetics 2008; 3: 1-4.
35. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008; 451: 1125-1129.
36. Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005; 123: 819-831.
37. Hirose J, Kouro T, Igarashi H, Yokota T, Sakaguchi N, Kincade PW. A developing picture of lymphopoiesis in bone marrow. Immunol Rev 2002; 189: 28-40.
38. Ciofani M, Zuniga-Pflucker JC. The thymus as an inductive site for T lymphopoiesis. Annu Rev Cell Dev Biol 2007; 23: 463-493.
39. O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 2010; 10: 111-122.
40. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007; 316: 608-611.
41. Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol 2008; 8: 380-390.
42. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 2004; 101: 2999-3004.
43. Vasilatou D, Papageorgiou S, Pappa V, Papageorgiou E, Dervenoulas J. The role of microRNAs in normal and malignant hematopoiesis. Eur J Haematol 2010; 84: 1-16.
44. Fabbri M, Croce CM, Calin GA. MicroRNAs in the ontogeny of leukemias and lymphomas. Leuk Lymphoma 2009; 50: 160-170.
45. Rego EM, Garcia AB, Carneiro JJ, Falcao RP. Immunophenotype of normal and leukemic bone marrow B-precursors in a Brazilian population. A comparative analysis by quantitative fluorescence cytometry. Braz J Med Biol Res 2001; 34: 183-194.
46. Rego EM, Tone LG, Garcia AB, Falcao RP. CD10 and CD19 fluorescence intensity of B-cell precursors in normal and leukemic bone marrow. Clinical characterization of CD10(+strong) and CD10(+weak) common acute lymphoblastic leukemia. Leuk Res 1999; 23: 441-450.
47. Schabath R, Ratei R, Ludwig WD. The prognostic significance of antigen expression in leukaemia. Best Pract Res Clin Haematol 2003; 16: 613-628.
48. Rego EM, Garcia AB, Viana SR, Falcao RP. Characterization of acute lymphoblastic leukemia subtypes in Brazilian patients. Leuk Res 1996; 20: 349-355.
49. Feltbower RG, McKinney PA, Greaves MF, Parslow RC, Bodansky HJ. International parallels in leukaemia and diabetes epidemiology. Arch Dis Child 2004; 89: 54-56.
50. Zanette DL, Rivadavia F, Molfetta GA, Barbuzano FG, Proto-Siqueira R, Silva-Jr WA, et al. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Braz J Med Biol Res 2007; 40: 1435-1440.
51. Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB, et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl Acad Sci U S A 2007; 104: 19971-19976.
52. Zhang H, Luo XQ, Zhang P, Huang LB, Zheng YS, Wu J, et al. MicroRNA patterns associated with clinical prognostic parameters and CNS relapse prediction in pediatric acute leukemia. PLoS One 2009; 4: e7826.
53. Kaddar T, Chien WW, Bertrand Y, Pages MP, Rouault JP, Salles G, et al. Prognostic value of miR-16 expression in childhood acute lymphoblastic leukemia relationships to normal and malignant lymphocyte proliferation. Leuk Res 2009; 33: 1217-1223.
54. Gimenes-Teixeira HL. Analysis of microRNA expression in T-cell acute lymphoblastic leukemia cells expressing or not the marker N-CAM (CD56). [Ph.D thesis]: Faculdade de Medicina de Ribeirão Preto, USP; 2009.
55. Dalmazzo LF, Jacomo RH, Marinato AF, Figueiredo-Pontes LL, Cunha RL, Garcia AB, et al. The presence of CD56/CD16 in T-cell acute lymphoblastic leukaemia correlates with the expression of cytotoxic molecules and is associated with worse response to treatment. Br J Haematol 2009; 144: 223-229.
56. Mi S, Li Z, Chen P, He C, Cao D, Elkahloun A, et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc Natl Acad Sci U S A 2010; 107: 3710-3715.
57. Li X, Liu J, Zhou R, Huang S, Huang S, Chen XM. Gene silencing of MIR22 in acute lymphoblastic leukaemia involves histone modifications independent of promoter DNA methylation. Br J Haematol 2010; 148: 69-79.
58. Tomita M, Tanaka Y, Mori N. MicroRNA miR-146a is induced by HTLV-1 tax and increases the growth of HTLV-1-infected T-cells. Int J Cancer 2009 (Epub ahead of print).
Address for correspondence: E.M. Rego, Divisão de Hematologia/Oncologia, Departamento de Clínica Médica, FMRP, USP, Av. Bandeirantes, 3900, 14049-900 Ribeirão Preto, SP, Brasil. E-mail: emrego@hcrp.fmrp.usp.br
Received April 26, 2010. Accepted May 25, 2010. Available online June 6, 2010. Published July 9, 2010.
The Brazilian Journal of Medical and Biological Research is partially financed by






- 1. Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol 2010; 220: 126-139.
- 2. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993; 75: 843-854.
- 3. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell 2009; 136: 642-655.
- 4. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans Nature 1998; 391: 806-811.
- 5. Mueller DW, Bosserhoff AK. Role of miRNAs in the progression of malignant melanoma. Br J Cancer 2009; 101: 551-556.
- 6. Shah PP, Hutchinson LE, Kakar SS. Emerging role of microRNAs in diagnosis and treatment of various diseases including ovarian cancer. J Ovarian Res 2009; 2: 11.
- 7. Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett 2009; 285: 116-126.
- 8. Spizzo R, Rushworth D, Guerrero M, Calin GA. RNA inhibition, microRNAs, and new therapeutic agents for cancer treatment. Clin Lymphoma Myeloma 2009; 9 (Suppl 3): S313-S318.
- 9. Corney DC, Nikitin AY. MicroRNA and ovarian cancer. Histol Histopathol 2008; 23: 1161-1169.
- 10. Croce CM. MicroRNAs and lymphomas. Ann Oncol 2008; 19 (Suppl 4): iv39-iv40.
- 11. Gomes CC, Gomez RS. MicroRNA and oral cancer: future perspectives. Oral Oncol 2008; 44: 910-914.
- 12. Nicoloso MS, Calin GA. MicroRNA involvement in brain tumors: from bench to bedside. Brain Pathol 2008; 18: 122-129.
- 13. Shi XB, Tepper CG, White RW. MicroRNAs and prostate cancer. J Cell Mol Med 2008; 12: 1456-1465.
- 14. Varnholt H. The role of microRNAs in primary liver cancer. Ann Hepatol 2008; 7: 104-113.
- 15. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23: 4051-4060.
- 16. Davis BN, Hata A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun Signal 2009; 7: 18.
- 17. Tsuchiya S, Okuno Y, Tsujimoto G. MicroRNA: biogenetic and functional mechanisms and involvements in cell differentiation and cancer. J Pharmacol Sci 2006; 101: 267-270.
- 18. Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007; 7: 105-117.
- 19. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004; 303: 83-86.
- 20. Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci U S A 2005; 102: 18081-18086.
- 21. Wang Q, Huang Z, Xue H, Jin C, Ju XL, Han JD, et al. MicroRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood 2008; 111: 588-595.
- 22. Dore LC, Amigo JD, dos Santos CO, Zhang Z, Gai X, Tobias JW, et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc Natl Acad Sci U S A 2008; 105: 3333-3338.
- 23. Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell 2008; 14: 843-853.
- 24. Kelly KF, Daniel JM. POZ for effect - POZ-ZF transcription factors in cancer and development. Trends Cell Biol 2006; 16: 578-587.
- 25. Labbaye C, Spinello I, Quaranta MT, Pelosi E, Pasquini L, Petrucci E, et al. A three-step pathway comprising PLZF/miR-146a/CXCR4 controls megakaryopoiesis. Nat Cell Biol 2008; 10: 788-801.
- 26. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006; 103: 12481-12486.
- 27. Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, et al. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 2007; 9: 775-787.
- 28. Rosa A, Ballarino M, Sorrentino A, Sthandier O, De Angelis FG, Marchioni M, et al. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc Natl Acad Sci U S A 2007; 104: 19849-19854.
- 29. Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol 2004; 339: 327-335.
- 30. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008; 132: 875-886.
- 31. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 2008; 9: 405-414.
- 32. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A 2007; 104: 1604-1609.
- 33. O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med 2008; 205: 585-594.
- 34. Nervi C, Fazi F, Grignani F. Oncoproteins, heterochromatin silencing and microRNAs: a new link for leukemogenesis. Epigenetics 2008; 3: 1-4.
- 35. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008; 451: 1125-1129.
- 36. Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005; 123: 819-831.
- 37. Hirose J, Kouro T, Igarashi H, Yokota T, Sakaguchi N, Kincade PW. A developing picture of lymphopoiesis in bone marrow. Immunol Rev 2002; 189: 28-40.
- 38. Ciofani M, Zuniga-Pflucker JC. The thymus as an inductive site for T lymphopoiesis. Annu Rev Cell Dev Biol 2007; 23: 463-493.
- 39. O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 2010; 10: 111-122.
- 40. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007; 316: 608-611.
- 41. Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol 2008; 8: 380-390.
- 42. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 2004; 101: 2999-3004.
- 43. Vasilatou D, Papageorgiou S, Pappa V, Papageorgiou E, Dervenoulas J. The role of microRNAs in normal and malignant hematopoiesis. Eur J Haematol 2010; 84: 1-16.
- 44. Fabbri M, Croce CM, Calin GA. MicroRNAs in the ontogeny of leukemias and lymphomas. Leuk Lymphoma 2009; 50: 160-170.
- 45. Rego EM, Garcia AB, Carneiro JJ, Falcao RP. Immunophenotype of normal and leukemic bone marrow B-precursors in a Brazilian population. A comparative analysis by quantitative fluorescence cytometry. Braz J Med Biol Res 2001; 34: 183-194.
- 46. Rego EM, Tone LG, Garcia AB, Falcao RP. CD10 and CD19 fluorescence intensity of B-cell precursors in normal and leukemic bone marrow. Clinical characterization of CD10(+strong) and CD10(+weak) common acute lymphoblastic leukemia. Leuk Res 1999; 23: 441-450.
- 47. Schabath R, Ratei R, Ludwig WD. The prognostic significance of antigen expression in leukaemia. Best Pract Res Clin Haematol 2003; 16: 613-628.
- 48. Rego EM, Garcia AB, Viana SR, Falcao RP. Characterization of acute lymphoblastic leukemia subtypes in Brazilian patients. Leuk Res 1996; 20: 349-355.
- 49. Feltbower RG, McKinney PA, Greaves MF, Parslow RC, Bodansky HJ. International parallels in leukaemia and diabetes epidemiology. Arch Dis Child 2004; 89: 54-56.
- 50. Zanette DL, Rivadavia F, Molfetta GA, Barbuzano FG, Proto-Siqueira R, Silva-Jr WA, et al. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Braz J Med Biol Res 2007; 40: 1435-1440.
- 51. Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB, et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl Acad Sci U S A 2007; 104: 19971-19976.
- 52. Zhang H, Luo XQ, Zhang P, Huang LB, Zheng YS, Wu J, et al. MicroRNA patterns associated with clinical prognostic parameters and CNS relapse prediction in pediatric acute leukemia. PLoS One 2009; 4: e7826.
- 53. Kaddar T, Chien WW, Bertrand Y, Pages MP, Rouault JP, Salles G, et al. Prognostic value of miR-16 expression in childhood acute lymphoblastic leukemia relationships to normal and malignant lymphocyte proliferation. Leuk Res 2009; 33: 1217-1223.
- 54. Gimenes-Teixeira HL. Analysis of microRNA expression in T-cell acute lymphoblastic leukemia cells expressing or not the marker N-CAM (CD56). [Ph.D thesis]: Faculdade de Medicina de Ribeirão Preto, USP; 2009.
- 55. Dalmazzo LF, Jacomo RH, Marinato AF, Figueiredo-Pontes LL, Cunha RL, Garcia AB, et al. The presence of CD56/CD16 in T-cell acute lymphoblastic leukaemia correlates with the expression of cytotoxic molecules and is associated with worse response to treatment. Br J Haematol 2009; 144: 223-229.
- 56. Mi S, Li Z, Chen P, He C, Cao D, Elkahloun A, et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc Natl Acad Sci U S A 2010; 107: 3710-3715.
- 57. Li X, Liu J, Zhou R, Huang S, Huang S, Chen XM. Gene silencing of MIR22 in acute lymphoblastic leukaemia involves histone modifications independent of promoter DNA methylation. Br J Haematol 2010; 148: 69-79.
- 58. Tomita M, Tanaka Y, Mori N. MicroRNA miR-146a is induced by HTLV-1 tax and increases the growth of HTLV-1-infected T-cells. Int J Cancer 2009 (Epub ahead of print).
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
05 July 2010 -
Date of issue
July 2010
History
-
Received
26 Apr 2010 -
Accepted
25 May 2010