Abstract
Andrographolide (ANDRO) has been studied for its immunomodulation, anti-inflammatory, and neuroprotection effects. Because brain hypoxia is the most common factor of secondary brain injury after traumatic brain injury, we studied the role and possible mechanism of ANDRO in this process using hypoxia-injured astrocytes. Mouse cortical astrocytes C8-D1A (astrocyte type I clone from C57/BL6 strains) were subjected to 3 and 21% of O2 for various times (0–12 h) to establish an astrocyte hypoxia injury model in vitro. After hypoxia and ANDRO administration, the changes in cell viability and apoptosis were assessed using CCK-8 and flow cytometry. Expression changes in apoptosis-related proteins, autophagy-related proteins, main factors of JNK pathway, ATG5, and S100B were determined by western blot. Hypoxia remarkably damaged C8-D1A cells evidenced by reduction of cell viability and induction of apoptosis. Hypoxia also induced autophagy and overproduction of S100B. ANDRO reduced cell apoptosis and promoted cell autophagy and S100B expression. After ANDRO administration, autophagy-related proteins, S-100B, JNK pathway proteins, and ATG5 were all upregulated, while autophagy-related proteins and s100b were downregulated when the jnk pathway was inhibited or ATG5 was knocked down. ANDRO conferred a survival advantage to hypoxia-injured astrocytes by reducing cell apoptosis and promoting autophagy and s100b expression. Furthermore, the promotion of autophagy and s100b expression by ANDRO was via activation of jnk pathway and regulation of ATG5.
Andrographolide; Hypoxia-injured astrocytes; JNK pathway; S100B; ATG5
Introduction
Traumatic brain injury (TBI) is a major cause of death or disability worldwide, especially in young people. After severe TBI, many people are killed by secondary injury, which is a complex set of cellular processes following trauma (11. Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ 2008; 178: 1163–1170, doi: 10.1503/cmaj.080282.
https://doi.org/10.1503/cmaj.080282...
). These secondary processes can worsen the damage and account for the greatest number of TBI deaths (22. Ghajar J. Traumatic brain injury. Kansas Nurse 2000; 356: 923–929.). Brain hypoxia is a common cause of secondary injury. Brain hypoxia initiates a sequence of biochemical events to induce neuronal cell apoptosis, resulting in a hypoxic brain injury and dysfunction (33. Yu ACH, Gregory GA, Chan PH. Hypoxia-Induced Dysfunctions and injury of astrocytes in primary cell cultures. J Cereb Blood Flow Metab 1989; 9: 20–28, doi: 10.1038/jcbfm.1989.3.
https://doi.org/10.1038/jcbfm.1989.3...
). Thus, brain hypoxia can aggravate TBI and is regarded as an independent predictor or a marker of disease severity (44. Oddo M, Levine JM, Mackenzie L, Frangos S, Feihl F, Kasner SE, et al. Brain hypoxia is associated with short-term outcome after severe traumatic brain injury independently of intracranial hypertension and low cerebral perfusion pressure. Neurosurgery 2011; 69: 1037, doi: 10.1227/NEU.0b013e3182287ca7.
https://doi.org/10.1227/NEU.0b013e318228...
).
Andrographis paniculata is a medicinal herb that is widely used in China and other parts of Asia for the treatment of upper respiratory tract infections, fever, and diarrhea (55. Coon JT, Ernst E. Andrographis paniculata in the treatment of upper respiratory tract infections: a systematic review of safety and efficacy. Planta Med 2004; 70: 293–298, doi: 10.1055/s-2004-818938.
https://doi.org/10.1055/s-2004-818938...
,66. Jarukamjorn K, Nemoto N. Pharmacological Aspects of Andrographis paniculata on Health and Its Major Diterpenoid Constituent Andrographolide. J Health Sci 2008; 54: 370–381, doi: 10.1248/jhs.54.370.
https://doi.org/10.1248/jhs.54.370...
). Andrographolide (ANDRO) is the major active component isolated from the stem and leaves of A. paniculata and is a natural diterpenoid lactone (77. Chakravarti RN, Chakravarti D. Andrographolide, the active constituent of Andrographis paniculata Nees; a preliminary communication. Ind Med Gaz 1951; 86: 96.). ANDRO has been studied for its various bioactivities including immunomodulation (88. Guan C, Min LI, Ren Q, Zhang W, Shan M, Wang R, et al. Andrographolide protects mice against severe Enterovirus 71 infection by its anti-inflammation and immunomodulation effects. Immunol J 2013; 29: 737–744.), anti-inflammatory (99. Chen HW, Lin AH, Chu HC, Li CC, Tsai CW, Chao CY, et al. Inhibition of TNF-α-Induced Inflammation by andrographolide via down-regulation of the PI3K/Akt signaling pathway. J Nat Prod 2011; 74: 2408–2413, doi: 10.1021/np200631v.
https://doi.org/10.1021/np200631v...
), anti-cancer (1010. Rajagopal S, Kumar RA, Deevi DS, Satyanarayana C, Rajagopalan R. Andrographolide, a potential cancer therapeutic agent isolated from Andrographis paniculata. J Exp Ther Oncol 2010; 3: 147–158, doi: 10.1046/j.1359-4117.2003.01090.x.
https://doi.org/10.1046/j.1359-4117.2003...
), anti-viral (1111. Lin TP, Chen SY, Duh PD, Chang LK, Liu YN. Inhibition of the epstein-barr virus lytic cycle by andrographolide. Biol Pharm Bull 2008; 31: 2018–2023, doi: 10.1248/bpb.31.2018.
https://doi.org/10.1248/bpb.31.2018...
), anti-bacterial (1212. Jiang X, Yu P, Jiang J, Zhang Z, Wang Z, Yang Z, et al. Synthesis and evaluation of antibacterial activities of andrographolide analogues. Eur J Medi Chem 2009; 44: 2936, doi: 10.1016/j.ejmech.2008.12.014.
https://doi.org/10.1016/j.ejmech.2008.12...
), anti-hyperglycemic (1313. Yu BC, Hung CR, Chen WC, Cheng JT. Antihyperglycemic effect of andrographolide in streptozotocin-induced diabetic rats. Planta Med 2003; 69: 1075–1079, doi: 10.1055/s-2003-45185.
https://doi.org/10.1055/s-2003-45185...
), and neuroprotective (1414. Chan SJ, Wong WF, Wong PT, Bian JS. Neuroprotective effects of andrographolide in a rat model of permanent cerebral ischaemia. Br J Pharmacol 2010; 161: 668–679, doi: 10.1111/j.1476-5381.2010.00906.x.
https://doi.org/10.1111/j.1476-5381.2010...
). The effect of ANDRO on cell apoptosis is complex. ANDRO protects thymocytes or endothelial cells against apoptosis (1515. Chen JH, Hsiao G, Lee AR, Wu CC, Yen MH. Andrographolide suppresses endothelial cell apoptosis via activation of phosphatidyl inositol-3-kinase/Akt pathway. Biochem Pharmacol 2004; 67: 1337–1345, doi: 10.1016/j.bcp.2003.12.015.
https://doi.org/10.1016/j.bcp.2003.12.01...
,1616. Burgos RA, Seguel K, Perez M, Meneses A, Ortega M, Guarda MI, et al. Andrographolide inhibits IFN-gamma and IL-2 cytokine production and protects against cell apoptosis. Planta Med 2005; 71: 429–434, doi: 10.1055/s-2005-864138.
https://doi.org/10.1055/s-2005-864138...
). On the other hand, some reports showed that ANDRO could induce apoptosis in human prostatic adenocarcinoma PC-3 cells (1717. Kim TG, Hwi KK, Hung CS. Morphological and biochemical changes of andrographolide-induced cell death in human prostatic adenocarcinoma PC-3 cells. In Vivo 2005; 19: 551–557.) or other cancer cells (1818. Zhao F, He EQ, Wang L, Liu K. Anti-tumor activities of andrographolide, a diterpene from Andrographis paniculata, by inducing apoptosis and inhibiting VEGF level. J Asian Nat Prod Res 2008; 10: 467–473, doi: 10.1080/10286020801948334.
https://doi.org/10.1080/1028602080194833...
). In addition, a previous study has showed that ANDRO could protect rat cardiomyocytes against hypoxia and reoxygenation injury (1919. Woo AYH, Waye MMY, Tsui SKW, Yeung STW, Cheng CHK. Andrographolide Up-Regulates Cellular-Reduced Glutathione Level and Protects Cardiomyocytes against Hypoxia/Reoxygenation Injury. J Pharmacol Exp Ther 2008; 325: 226–235, doi: 10.1124/jpet.107.133918.
https://doi.org/10.1124/jpet.107.133918...
). Furthermore, ANDRO was reported to protect mice against hypoxia/ischemia-induced oxidative brain injury (2020. Chern CM, Liou KT, Wang YH, Liao JF, Yen JC, Shen YC. Andrographolide inhibits PI3K/AKT-dependent NOX2 and iNOS expression protecting mice against hypoxia/ischemia-induced oxidative brain injury. Planta Med 2011; 77: 1669–1679, doi: 10.1055/s-0030-1271019.
https://doi.org/10.1055/s-0030-1271019...
). Therefore, we aimed to investigate whether ANDRO has an effect on mouse hypoxia-injured brain cells, which is still unknown.
Astrocytes are the most widely distributed class of cells in mammalian brain. These cells perform many functions, including support of endothelial cells, fueling neurons, maintenance of extracellular ion balance, nervous repair, and scarring of the brain following traumatic injuries (2121. Harder DR, Zhang C, Gebremedhin D. Astrocytes function in matching blood flow to metabolic activity. Physiol 2002; 17: 27–31, doi: 10.1152/physiologyonline.2002.17.1.27.
https://doi.org/10.1152/physiologyonline...
22. Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016; 532: 195, doi: 10.1038/nature17623.
https://doi.org/10.1038/nature17623...
–2323. Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 2000; 36: 291–300, doi: 10.1016/S0197-0186(99)00137-0.
https://doi.org/10.1016/S0197-0186(99)00...
). Therefore, we constructed a model of hypoxia injury in mouse astrocytes to investigate the role of ANDRO. To achieve this goal, we revealed the role of ANDRO from three aspects: cell apoptosis, autophagy, and S100B expression.
Material and Methods
Cell culture
Mouse cortical astrocytes C8-D1A (astrocyte type I clone from C57/BL6 strains), were purchased from the American Type Culture Collection (ATCC, CRL-2541TM, USA). Cells were cultured with high glucose Dulbecco’s modified eagle medium (DMEM, Invitrogen, CA) containing 10% fetal bovine serum (FBS, Gibco, USA), 50 IU/mL penicillin G (Gibco, USA) and 50 μg/mL streptomycin (Gibco). Then, cells were incubated at 37°C in a moist atmosphere containing 5% CO2.
Hypoxia exposure
To establish an astrocytes hypoxia injury model, cells were subjected to hypoxic (3% O2, 5% CO2 balanced with N2) culture. Cells cultured in normoxia (room air, 5% CO2) were the control.
ANDRO treatment
ANDRO was purchased from Sigma-Aldrich (365645, USA) with purity ≥98%. A stock solution with a concentration of 15 mM was prepared by dissolving ANDRO in dimethyl sulphoxide (Sigma-Aldrich, USA). ANDRO in a concentration of 10 μM was used to treat cells throughout the hypoxia exposure. SP600125, an inhibitor of JNK, was purchased from Sigma (S5567, USA). SP600125 in a concentration of 10 μM was used to treat cells throughout the hypoxia exposure.
Viability assay
Cells were seeded on a 96-well plate with 5000 cells/well. Cell viability was measured by the Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, USA). After incubation for 0–12 h at 37°C, cells from control, hypoxia, and hypoxia (12 h)+ANDRO groups were added to CCK-8 solution with 10 μL/well and were incubated for 4 h at 37°C. Then, the absorbance was measured at 450 nM using a Microplate Reader (Bio-Rad, USA).
Apoptosis assay
Cells for apoptosis detection were washed twice with cold PBS and resuspended in buffer. Then, cells were dyed using Annexin V-FITC/PI apoptosis detection kit (Invitrogen, USA) according to the manufacturer’s instruction. After reaction in dark at room temperature for 10 min, flow cytometry analysis was done by using a FACScan flow cytometer (Beckton Dickinson, USA) to differentiate apoptotic cells (Annexin-V positive and PI negative) from necrotic cells (Annexin-V and PI positive).
Cell transfection
siRNA targeted against ATG5 (si-ATG5) and non-targeting siRNA (si-NC) used as negative control were synthesized from GenePharma Co. (China). Cell transfections were conducted using Lipofectamine 3000 reagent (Invitrogen, USA) following the manufacturer’s protocol. After 48 h of transfection, cells were collected for further analysis.
Real-time quantitative reverse transcriptase PCR (qRT-PCR)
Total RNA was isolated from treated cells by using TRIzol reagent (Invitrogen, USA). Reverse transcription was performed by using the Prime Script RT reagent Kit (TaKaRa, China) according to the manufacturer’s instructions. A SYBR Fast qPCR Mix (TaKaRa) was used to quantify the mRNA levels according to the manufacturer’s instructions. β-actin was used as the internal standard. The ΔΔCt method was used to calculate changes in expression.
Western blot
The proteins used for western blot were extracted using RIPA lysis and extraction buffer (Thermo Scientific, USA). The proteins were quantified using the BCA Protein Assay Kit (Thermo Scientific) and were separated by a NativePAGE Novex Bis-Tris Gel system (Invitrogen) according to the manufacturer’s instructions. Then proteins were transferred to a Polyvinylidene Difluoride (PVDF) membrane (Millipore, USA). The membrane was incubated with primary antibodies at 4°C overnight, followed by washing and incubation with secondary antibody marked by horseradish peroxidase at room temperature. Primary antibodies included anti-Bcl-2 (ab59348, 1:1000; Abcam, UK), anti-Bax (ab182733, 1:2000; Abcam), anti-pro-caspase-3 (ab44976, 1:500; Abcam), anti-cleaved-caspase-3 (ab13847, 1:500; Abcam), anti-caspase-9 (ab202068, 1:2000; Abcam), anti-LC3B (ab51520, 1:3000, Abcam), anti-Beclin-1 (ab62557, 1:1000; Abcam), anti-p62 (ab56416, 1:1000; Abcam), anti-β-actin (ab8224, 1:1000; Abcam), anti-S100B (ab52642, 1:1000; Abcam), anti-JNK (ab179461, 1:1000; Abcam), anti-p-JNK (ab124956, 1:5000; Abcam), anti-c-Jun (ab32137, 1:5000; Abcam), anti-p-c-Jun (ab32385, 1:5000; Abcam), and anti-ATG5 (ab108327, 1:5000; Abcam). After adding ECL Plus Western Blotting Substrate (Thermo Scientific) to cover the membrane surface, the signals were captured and the intensity of the bands was quantified using the ChemiDoc™ XRS system (Bio-Rad, USA). β-actin antibody (Abcam) was used as the endogenous protein for reference.
Statistical analysis
All experiments were repeated three times. The results of multiple experiments are reported as means±SD. Statistical analyses were performed using SPSS 22.0 statistical software (SPSS, USA) using a one-way analysis of variance (ANOVA). A P-value of <0.05 indicates a statistically significant result.
Results
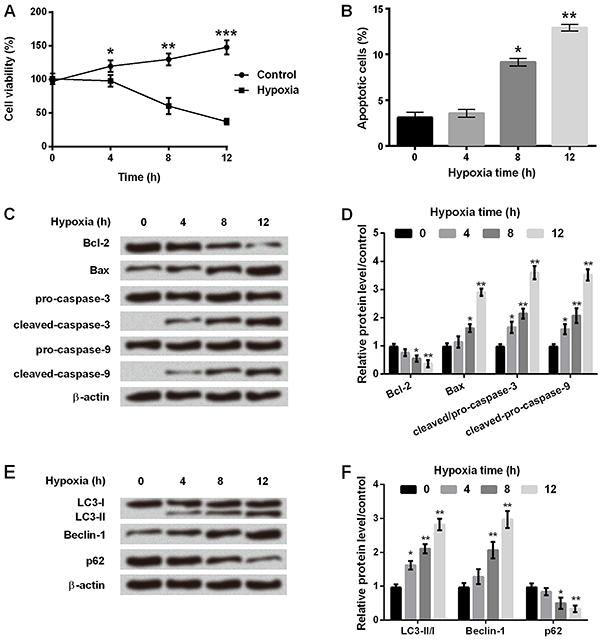
Hypoxia affected cell viability, apoptosis, and autophagy of astrocytes
After culturing in hypoxia or control (normoxia) environment for 0 to 12 h, C8-D1A cells were harvested and analyzed for cell viability, apoptosis, and autophagy. As shown in Figure 1A, cell viability was significantly reduced at 4 to 12 h (P<0.05, P<0.01 or P<0.001) after hypoxia. Flow cytometry analysis demonstrated that the number of apoptotic cells was significantly increased after 8 h (P<0.05) and 12 h (P<0.01) in hypoxia (Figure 1B). Figure 1C and D showed that Bcl-2 expression was significantly decreased, while Bax, cleaved-caspase-3, and cleaved-caspase-9 expressions were significantly increased after 8 h in hypoxia incubator compared with control (P<0.05 or P<0.01). We also analyzed expression of proteins involved in cell autophagy, and found that the ratio of LC3-II/LC-I and the amount of Beclin-1 were significantly increased, while that of p62 was decreased in a time-dependent manner in hypoxia incubator compared to control (Figure 1E and F, P<0.05 or P<0.01). These results suggested that cell apoptosis and autophagy were significantly increased after 8 h of hypoxic incubation in astrocytes.
A, Cell viability was determined at different times in the control and hypoxia groups. B, Apoptosis rate was measured at different times in hypoxic astrocytes. C, Protein immunoblots of apoptosis-related factors by western blot assay. D, Relative expressions of apoptosis-related factors by quantification of band intensity. E, Protein immunoblots of autophagy-related factors by western blot assay. F, Relative expressions of autophagy-related factors by quantification of band intensity. β-actin acted as an internal control. Data are reported as means±SD. *P<0.05, **P<0.01, ***P<0.001 compared to 0 h (ANOVA).
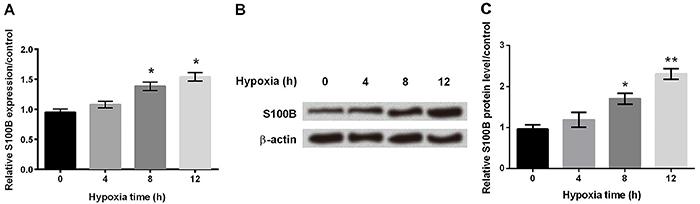
Hypoxia induced S100B expression in astrocytes
Using qRT-PCR, we demonstrated that the mRNA amount of S100B was significantly increased after 8 h in hypoxia (Figure 2A, P<0.05). Western blot also confirmed that protein expression of S100B was gradually increased in hypoxic-treated astrocytes (Figure 2B and C, P<0.05 or P<0.01).
A, mRNA expression of S100B was determined by qRT-PCR at different times in hypoxic astrocytes. B and C, Western blot was performed to assess the protein expression of S100B at different times in hypoxia astrocytes. β-actin acted as an internal control. Data are reported as means±SD. *P<0.05, **P<0.01 compared to 0 h (ANOVA).
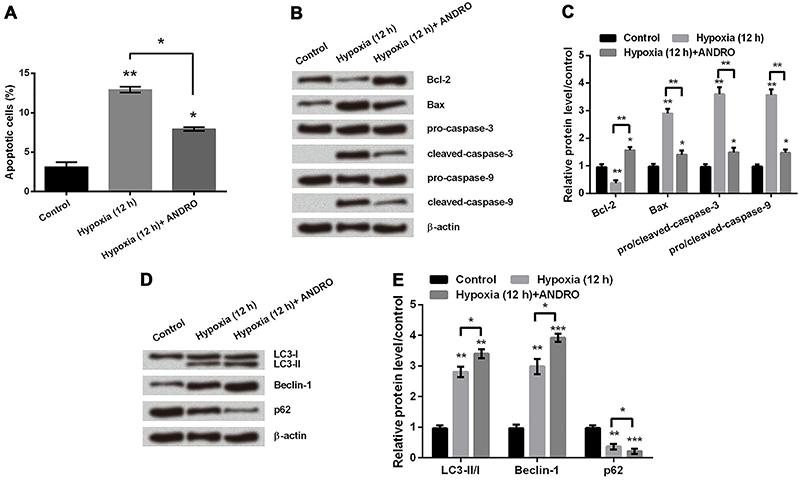
ANDRO reduced cell apoptosis but promoted cell autophagy in hypoxic astrocytes
To reveal the effect of ANDRO on apoptosis, firstly we used flow cytometry analysis. The number of apoptotic cells in hypoxic astrocytes was significantly decreased after adding ANDRO (Figure 3A, P<0.05). Secondly, we detected expression levels of proteins involved in apoptosis by western blot. As shown in Figure 3B and C, addition of ANDRO in hypoxic astrocytes resulted in an increase in Bcl-2 protein expression and decreases in Bax, cleaved-Caspase-3, and cleaved-Caspase-9 protein expressions (P<0.05 or P<0.01). These results suggested that ANDRO attenuates the process of apoptosis in hypoxic astrocytes. By analyzing expression of proteins related to cell autophagy after 12 h in hypoxia, we found that addition of ANDRO significantly promoted increases in LC3-II and Beclin-1 protein expressions, and a decline in p62 protein expression (Figure 3D and E, P<0.05, P<0.01 or P<0.001). This data showed that ANDRO promoted autophagy in hypoxic astrocytes.
A, Apoptosis rate was measured in hypoxic astrocytes with or without treatment of andrographolide (ANDRO). B and C, Western blot was used to determine the protein expression level of apoptosis related proteins in hypoxic astrocytes with or without treatment of ANDRO. D and E, Protein expression of autophagy-regulated factors was determined by western blot. β-actin acted as an internal control. Data are reported as means±SD. *P<0.05, **P<0.01, ***P<0.001 (ANOVA).
ANDRO promoted upregulation of S100B expression
Both qRT-PCR (Figure 4A) and western blot analysis (Figure 4B and C) suggested that addition of ANDRO dramatically increased the expression of S100B in hypoxic astrocytes (P<0.05 or P<0.01).
A, qRT-PCR was used to determine the mRNA expression of S100B in hypoxic astrocytes with or without treatment of andrographolide (ANDRO). B and C, Western blot was performed to assess the protein expression of S100B in hypoxic astrocytes with or without treatment of ANDRO. β-actin acted as an internal control. Data are reported as means±SD. *P<0.05, **P<0.01 (ANOVA).
ANDRO promoted autophagy and S100B expression by activating JNK signaling pathways
Next, we investigated the underlying mechanisms through which ANDRO promoted autophagy and expression of S100B. As shown in Figure 5A and B, adding ANDRO alone promoted expressions of S100B and autophagy-related proteins as described above. In addition, ANDRO increased the expressions of p-JNK and p-c-Jun. However, after treatment with JNK inhibitor, ANDRO treatment did not lead to increases in the expressions of LC3-II, Beclin-1, and S100B, or a decline in p62 expression. These results suggested that the promotion of autophagy and S100B expression by ANDRO might be through activating JNK signaling pathways in hypoxic astrocytes.
A, Protein immunoblots of S100B and proteins related to JNK pathway and autophagy measured by western blot in hypoxic astrocytes with or without treatment of andrographolide (ANDRO) and SP600125. B, Relative expressions of JNK pathway and autophagy related factors by quantification of band intensity. β-actin acted as an internal control. Data are reported as means±SD. *P<0.05, **P<0.01, ***P<0.001 (ANOVA).
ANDRO promoted autophagy and S100B expression by upregulating expression of ATG5
To reveal the regulation mechanism of ANDRO, we also demonstrated the association between ANDRO and expressions of S100B and ATG5. Firstly, we found that expression of ATG5 was increased in hypoxic astrocytes with the treatment of ANDRO (Figure 6A and B, P<0.05, P<0.01 or P<0.001). Given the important role of ATG5 in autophagy process (2424. Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009; 6: 354–366, doi: 10.1016/j.chom.2009.09.006.
https://doi.org/10.1016/j.chom.2009.09.0...
), this result provided more evidence that ANDRO promoted autophagy in hypoxic astrocytes. Secondly, when ATG5 was knocked down in hypoxic astrocytes with ANDRO treatment, expression of autophagy-related proteins (LC3-II, Beclin-1, and p62) were reduced, and interestingly the expression level of S100B was also reduced to that in control cells (P<0.05, P<0.01 or P<0.001). Therefore, we concluded that ANDRO promoted S100B expression perhaps by upregulating expression of ATG5.
A, Protein immunoblots of ATG5, S100B, and autophagy-related proteins measured by western blot in hypoxic astrocytes with or without si-ATG5 transfection and treatment of andrographolide (ANDRO). B, Relative expressions of ATG5, S100B and autophagy-related proteins by quantification of band intensity. β-actin acted as an internal control. si-NC: non-targeting siRNA. Data are reported as means±SD. *P<0.05, **P<0.01, ***P<0.001 (ANOVA).
Discussion
ANDRO has been well known for its various bioactivities such as anti-inflammatory (99. Chen HW, Lin AH, Chu HC, Li CC, Tsai CW, Chao CY, et al. Inhibition of TNF-α-Induced Inflammation by andrographolide via down-regulation of the PI3K/Akt signaling pathway. J Nat Prod 2011; 74: 2408–2413, doi: 10.1021/np200631v.
https://doi.org/10.1021/np200631v...
). However, in hypoxia brain injury, the function of ANDRO has not been well studied. Recently ANDRO was reported to protect rat cardiomyocytes against hypoxia and reoxygenation injury and protect mice brain against hypoxia/ischemia injury (1919. Woo AYH, Waye MMY, Tsui SKW, Yeung STW, Cheng CHK. Andrographolide Up-Regulates Cellular-Reduced Glutathione Level and Protects Cardiomyocytes against Hypoxia/Reoxygenation Injury. J Pharmacol Exp Ther 2008; 325: 226–235, doi: 10.1124/jpet.107.133918.
https://doi.org/10.1124/jpet.107.133918...
,2020. Chern CM, Liou KT, Wang YH, Liao JF, Yen JC, Shen YC. Andrographolide inhibits PI3K/AKT-dependent NOX2 and iNOS expression protecting mice against hypoxia/ischemia-induced oxidative brain injury. Planta Med 2011; 77: 1669–1679, doi: 10.1055/s-0030-1271019.
https://doi.org/10.1055/s-0030-1271019...
). Similarly, in the present study, we demonstrated that ANDRO protected mouse astrocytes against hypoxia injury.
To research the role of ANDRO in hypoxia-injured astrocytes, we investigated its effects on three aspects: cell apoptosis, autophagy, and S100B expression. Flow cytometry analysis was performed to detect apoptotic cell rate, and expressions of a series of apoptosis-related proteins were measured. Caspases play an essential role in the transduction of apoptotic signals. When cytochrome c is released into the cytosol, it binds to an adaptor protein and pro-caspase-9, which in turn cleaves the pro-caspase-9 into the active form (2525. Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci 2010; 123: 3209–3214, doi: 10.1242/jcs.073643.
https://doi.org/10.1242/jcs.073643...
). Caspase-3 is activated by cleaved-caspase-9 through proteolytic cleavage; then it degrades many intracellular proteins to carry out programed cell death (2626. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 2004; 5: 897–907, doi: 10.1038/nrm1496.
https://doi.org/10.1038/nrm1496...
). The Bcl-2 family, including anti-apoptotic (Bcl-2, Bcl-XL, Mcl-1) and pro-apoptotic members (Bid, Bax, Bad), is also one of the apoptotic regulatory proteins (2727. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647, doi: 10.1038/nrc883.
https://doi.org/10.1038/nrc883...
). Therefore, we used cleaved-caspase-3, cleaved-caspase-9, Bcl-2, and Bax as markers of apoptosis in our study. As a result, we found that the apoptotic cell rate and pro-apoptosis-related protein expressions were decreased and anti-apoptosis-related protein expression was increased by ANDRO, indicating that it attenuated the process of apoptosis in hypoxia-injured astrocytes.
Autophagy, also known as type II programmed cell death, causes orderly degradation and recycling of cellular components to survive bioenergetic stress (2828. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147: 728–741, doi: 10.1016/j.cell.2011.10.026.
https://doi.org/10.1016/j.cell.2011.10.0...
). Beclin-1 is a component of the phosphatidylinositol-3-kinase complex that is required for autophagy (2929. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19: 5360–5372, doi: 10.1091/mbc.E08-01-0080.
https://doi.org/10.1091/mbc.E08-01-0080...
). Beclin-1 expression has been recently used as a marker of autophagy (3030. Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis 2008; 29: 132–141, doi: 10.1016/j.nbd.2007.08.005.
https://doi.org/10.1016/j.nbd.2007.08.00...
,3131. Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 2008; 32: 329–339, doi: 10.1016/j.nbd.2008.07.022.
https://doi.org/10.1016/j.nbd.2008.07.02...
). During autophagy, there are two ATG5-dependent ubiquitin-like conjugation systems: the ATG12 conjugation system, leading to the formation of ATG12-ATG5-ATG16 molecular complexes, and the LC3 conjugation system, causing the LC3-I to generate a lipidated LC3-II form (2424. Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009; 6: 354–366, doi: 10.1016/j.chom.2009.09.006.
https://doi.org/10.1016/j.chom.2009.09.0...
). LC3 also enables the docking of specific cargos and adaptor proteins such as p62 (Sequestosome-1)(3232. Sungwoo P, Seon-Guk C, Seung-Min Y, Jin H, Son, Yong-Keun J. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014; 10: 1906–1920, doi: 10.4161/auto.32177.
https://doi.org/10.4161/auto.32177...
). Both conjugation systems lead to the formation of autophagosomes (2424. Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009; 6: 354–366, doi: 10.1016/j.chom.2009.09.006.
https://doi.org/10.1016/j.chom.2009.09.0...
). Therefore, we used Beclin-1, LC3II, and p62 as markers of autophagy in our experiments. Results showed that expressions of Beclin-1 and LC3II were increased and p62 was decreased by ANDRO. Therefore, we concluded that ANDRO promoted autophagy in hypoxia-injured astrocytes. Many studies have shown that autophagy plays a protective role in brain injury (3232. Sungwoo P, Seon-Guk C, Seung-Min Y, Jin H, Son, Yong-Keun J. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014; 10: 1906–1920, doi: 10.4161/auto.32177.
https://doi.org/10.4161/auto.32177...
,3333. Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med 2012; 25 (Suppl 1): 30, doi: 10.3109/14767058.2012.663176.
https://doi.org/10.3109/14767058.2012.66...
), and our results also confirm this point. Considering that ANDRO inhibited apoptosis while promoting autophagy, we can conclude that ANDRO induced cell autophagy from apoptosis to protect astrocytes from hypoxia damage.
S100B, produced mainly by astrocytes, exerts a neurotrophic effect and was shown to reduce brain injury (3434. Willoughby KA, Kleindienst A, Müller C, Chen T, Muir JK, Ellis EF. S100B protein is released by in vitro trauma and reduces delayed neuronal injury. J Neurochem 2010; 91: 1284–1291, doi: 10.1111/j.1471-4159.2004.02812.x.
https://doi.org/10.1111/j.1471-4159.2004...
,3535. Ellis EF, Willoughby KA, Sparks SA, Chen T. S100B protein is released from rat neonatal neurons, astrocytes, and microglia by invitro trauma and anti-S100 increases trauma-induced delayed neuronal injury and negates the protective effect of exogenous S100B on neurons. J Neurochem 2007; 101: 1463–1470, doi: 10.1111/j.1471-4159.2007.04515.x.
https://doi.org/10.1111/j.1471-4159.2007...
). In the present study, we found that the expression of S100B was increased by ANDRO in hypoxia-injured astrocytes. Based on these findings, we inferred that ANDRO protected astrocytes against hypoxia injury by promoting S100B expression.
Next, in order to determine the mechanism responsible for regulation of autophagy and S100B expression by ANDRO, we tested the association between ANDRO, autophagy, and expression levels of S100B, JNK pathway proteins, and ATG5. Activated JNK regulates several important cellular functions including cell growth, differentiation, survival, and apoptosis by activating some small molecules such as c-Jun (3636. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000; 103: 239–252, doi: 10.1016/S0092-8674(00)00116-1.
https://doi.org/10.1016/S0092-8674(00)00...
). Recently, JNK has been found to regulate autophagy (3737. Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy 2010; 6: 153, doi: 10.4161/auto.6.1.10537.
https://doi.org/10.4161/auto.6.1.10537...
,3838. Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med 2011; 51: 1365–1375, doi: 10.1016/j.freeradbiomed.2011.06.016.
https://doi.org/10.1016/j.freeradbiomed....
). In the current study, we found that ANDRO activated JNK pathway. When JNK pathway was inhibited, ANDRO could not upregulate autophagy and S100B expression as before. These results indicated that the promoting activities of ANDRO on autophagy and S100B expression might be through the JNK pathway. On the other hand, our study showed that knocking down ATG5 resulted in reduction of S100B expression in hypoxia-injured astrocytes with ANDRO treatment. This indicated that ATG5 reduced the expression of S100B even under the action of ANDRO. We, therefore, concluded that ANDRO promoted autophagy via the increased expression of S100B. However, this needs further research.
In conclusion, the present study indicated that ANDRO protected hypoxia-injured astrocytes against apoptosis and promoted autophagy and S100B expression. Our findings may have important implications in the treatment of hypoxia brain injury and application of ANDRO, although further research is still needed.
References
-
1Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ 2008; 178: 1163–1170, doi: 10.1503/cmaj.080282.
» https://doi.org/10.1503/cmaj.080282 -
2Ghajar J. Traumatic brain injury. Kansas Nurse 2000; 356: 923–929.
-
3Yu ACH, Gregory GA, Chan PH. Hypoxia-Induced Dysfunctions and injury of astrocytes in primary cell cultures. J Cereb Blood Flow Metab 1989; 9: 20–28, doi: 10.1038/jcbfm.1989.3.
» https://doi.org/10.1038/jcbfm.1989.3 -
4Oddo M, Levine JM, Mackenzie L, Frangos S, Feihl F, Kasner SE, et al. Brain hypoxia is associated with short-term outcome after severe traumatic brain injury independently of intracranial hypertension and low cerebral perfusion pressure. Neurosurgery 2011; 69: 1037, doi: 10.1227/NEU.0b013e3182287ca7.
» https://doi.org/10.1227/NEU.0b013e3182287ca7 -
5Coon JT, Ernst E. Andrographis paniculata in the treatment of upper respiratory tract infections: a systematic review of safety and efficacy. Planta Med 2004; 70: 293–298, doi: 10.1055/s-2004-818938.
» https://doi.org/10.1055/s-2004-818938 -
6Jarukamjorn K, Nemoto N. Pharmacological Aspects of Andrographis paniculata on Health and Its Major Diterpenoid Constituent Andrographolide. J Health Sci 2008; 54: 370–381, doi: 10.1248/jhs.54.370.
» https://doi.org/10.1248/jhs.54.370 -
7Chakravarti RN, Chakravarti D. Andrographolide, the active constituent of Andrographis paniculata Nees; a preliminary communication. Ind Med Gaz 1951; 86: 96.
-
8Guan C, Min LI, Ren Q, Zhang W, Shan M, Wang R, et al. Andrographolide protects mice against severe Enterovirus 71 infection by its anti-inflammation and immunomodulation effects. Immunol J 2013; 29: 737–744.
-
9Chen HW, Lin AH, Chu HC, Li CC, Tsai CW, Chao CY, et al. Inhibition of TNF-α-Induced Inflammation by andrographolide via down-regulation of the PI3K/Akt signaling pathway. J Nat Prod 2011; 74: 2408–2413, doi: 10.1021/np200631v.
» https://doi.org/10.1021/np200631v -
10Rajagopal S, Kumar RA, Deevi DS, Satyanarayana C, Rajagopalan R. Andrographolide, a potential cancer therapeutic agent isolated from Andrographis paniculata. J Exp Ther Oncol 2010; 3: 147–158, doi: 10.1046/j.1359-4117.2003.01090.x.
» https://doi.org/10.1046/j.1359-4117.2003.01090.x -
11Lin TP, Chen SY, Duh PD, Chang LK, Liu YN. Inhibition of the epstein-barr virus lytic cycle by andrographolide. Biol Pharm Bull 2008; 31: 2018–2023, doi: 10.1248/bpb.31.2018.
» https://doi.org/10.1248/bpb.31.2018 -
12Jiang X, Yu P, Jiang J, Zhang Z, Wang Z, Yang Z, et al. Synthesis and evaluation of antibacterial activities of andrographolide analogues. Eur J Medi Chem 2009; 44: 2936, doi: 10.1016/j.ejmech.2008.12.014.
» https://doi.org/10.1016/j.ejmech.2008.12.014 -
13Yu BC, Hung CR, Chen WC, Cheng JT. Antihyperglycemic effect of andrographolide in streptozotocin-induced diabetic rats. Planta Med 2003; 69: 1075–1079, doi: 10.1055/s-2003-45185.
» https://doi.org/10.1055/s-2003-45185 -
14Chan SJ, Wong WF, Wong PT, Bian JS. Neuroprotective effects of andrographolide in a rat model of permanent cerebral ischaemia. Br J Pharmacol 2010; 161: 668–679, doi: 10.1111/j.1476-5381.2010.00906.x.
» https://doi.org/10.1111/j.1476-5381.2010.00906.x -
15Chen JH, Hsiao G, Lee AR, Wu CC, Yen MH. Andrographolide suppresses endothelial cell apoptosis via activation of phosphatidyl inositol-3-kinase/Akt pathway. Biochem Pharmacol 2004; 67: 1337–1345, doi: 10.1016/j.bcp.2003.12.015.
» https://doi.org/10.1016/j.bcp.2003.12.015 -
16Burgos RA, Seguel K, Perez M, Meneses A, Ortega M, Guarda MI, et al. Andrographolide inhibits IFN-gamma and IL-2 cytokine production and protects against cell apoptosis. Planta Med 2005; 71: 429–434, doi: 10.1055/s-2005-864138.
» https://doi.org/10.1055/s-2005-864138 -
17Kim TG, Hwi KK, Hung CS. Morphological and biochemical changes of andrographolide-induced cell death in human prostatic adenocarcinoma PC-3 cells. In Vivo 2005; 19: 551–557.
-
18Zhao F, He EQ, Wang L, Liu K. Anti-tumor activities of andrographolide, a diterpene from Andrographis paniculata, by inducing apoptosis and inhibiting VEGF level. J Asian Nat Prod Res 2008; 10: 467–473, doi: 10.1080/10286020801948334.
» https://doi.org/10.1080/10286020801948334 -
19Woo AYH, Waye MMY, Tsui SKW, Yeung STW, Cheng CHK. Andrographolide Up-Regulates Cellular-Reduced Glutathione Level and Protects Cardiomyocytes against Hypoxia/Reoxygenation Injury. J Pharmacol Exp Ther 2008; 325: 226–235, doi: 10.1124/jpet.107.133918.
» https://doi.org/10.1124/jpet.107.133918 -
20Chern CM, Liou KT, Wang YH, Liao JF, Yen JC, Shen YC. Andrographolide inhibits PI3K/AKT-dependent NOX2 and iNOS expression protecting mice against hypoxia/ischemia-induced oxidative brain injury. Planta Med 2011; 77: 1669–1679, doi: 10.1055/s-0030-1271019.
» https://doi.org/10.1055/s-0030-1271019 -
21Harder DR, Zhang C, Gebremedhin D. Astrocytes function in matching blood flow to metabolic activity. Physiol 2002; 17: 27–31, doi: 10.1152/physiologyonline.2002.17.1.27.
» https://doi.org/10.1152/physiologyonline.2002.17.1.27 -
22Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016; 532: 195, doi: 10.1038/nature17623.
» https://doi.org/10.1038/nature17623 -
23Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 2000; 36: 291–300, doi: 10.1016/S0197-0186(99)00137-0.
» https://doi.org/10.1016/S0197-0186(99)00137-0 -
24Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009; 6: 354–366, doi: 10.1016/j.chom.2009.09.006.
» https://doi.org/10.1016/j.chom.2009.09.006 -
25Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci 2010; 123: 3209–3214, doi: 10.1242/jcs.073643.
» https://doi.org/10.1242/jcs.073643 -
26Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 2004; 5: 897–907, doi: 10.1038/nrm1496.
» https://doi.org/10.1038/nrm1496 -
27Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647, doi: 10.1038/nrc883.
» https://doi.org/10.1038/nrc883 -
28Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147: 728–741, doi: 10.1016/j.cell.2011.10.026.
» https://doi.org/10.1016/j.cell.2011.10.026 -
29Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19: 5360–5372, doi: 10.1091/mbc.E08-01-0080.
» https://doi.org/10.1091/mbc.E08-01-0080 -
30Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis 2008; 29: 132–141, doi: 10.1016/j.nbd.2007.08.005.
» https://doi.org/10.1016/j.nbd.2007.08.005 -
31Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 2008; 32: 329–339, doi: 10.1016/j.nbd.2008.07.022.
» https://doi.org/10.1016/j.nbd.2008.07.022 -
32Sungwoo P, Seon-Guk C, Seung-Min Y, Jin H, Son, Yong-Keun J. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014; 10: 1906–1920, doi: 10.4161/auto.32177.
» https://doi.org/10.4161/auto.32177 -
33Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med 2012; 25 (Suppl 1): 30, doi: 10.3109/14767058.2012.663176.
» https://doi.org/10.3109/14767058.2012.663176 -
34Willoughby KA, Kleindienst A, Müller C, Chen T, Muir JK, Ellis EF. S100B protein is released by in vitro trauma and reduces delayed neuronal injury. J Neurochem 2010; 91: 1284–1291, doi: 10.1111/j.1471-4159.2004.02812.x.
» https://doi.org/10.1111/j.1471-4159.2004.02812.x -
35Ellis EF, Willoughby KA, Sparks SA, Chen T. S100B protein is released from rat neonatal neurons, astrocytes, and microglia by invitro trauma and anti-S100 increases trauma-induced delayed neuronal injury and negates the protective effect of exogenous S100B on neurons. J Neurochem 2007; 101: 1463–1470, doi: 10.1111/j.1471-4159.2007.04515.x.
» https://doi.org/10.1111/j.1471-4159.2007.04515.x -
36Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000; 103: 239–252, doi: 10.1016/S0092-8674(00)00116-1.
» https://doi.org/10.1016/S0092-8674(00)00116-1 -
37Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy 2010; 6: 153, doi: 10.4161/auto.6.1.10537.
» https://doi.org/10.4161/auto.6.1.10537 -
38Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med 2011; 51: 1365–1375, doi: 10.1016/j.freeradbiomed.2011.06.016.
» https://doi.org/10.1016/j.freeradbiomed.2011.06.016
Publication Dates
-
Publication in this collection
2018
History
-
Received
26 Aug 2017 -
Accepted
21 Dec 2017