Abstract
Reactive oxygen species (ROS) are highly reactive chemical species that may cause irreversible tissue damage, and play a critical role in cardiovascular diseases. Hydrogen sulfide (H2S) is a gasotransmitter that acts as a ROS scavenger with cardio-protective effects. In this study, we investigated the cytoprotective effect of H2S against H2O2-induced apoptosis in cardiomyocytes. H9c2 rat cardiomyoblasts were treated with H2S (100 μM) 24 h before challenging with H2O2 (100 μM). Apoptosis was then assessed by annexin V and PI, and mitochondrial membrane potential was measured using a fluorescent probe, JC-1. Our results revealed that H2S improved cell viability, reduced the apoptotic rate, and preserved mitochondrial membrane potential. An increased Bcl-2 to Bax ratio was also seen in myocytes treated with H2S after H2O2-induced stress. Our findings indicated a therapeutic potential for H2S in preventing myocyte death following ischemia/reperfusion.
Reactive oxygen species; ROS; Hydrogen sulfide; H2O2-induced apoptosis; Cardiomyocytes
Introduction
Reactive oxygen species (ROS) are highly reactive chemical species from cellular metabolism involving oxygen consumption (11. Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med 2000; 109: 315–323, doi: 10.1016/S0002-9343(00)00467-8.
https://doi.org/10.1016/S0002-9343(00)00...
). In normal tissues, approximately 5% of the consumed oxygen molecules are transformed into ROS (22. McCord JM. Free radicals and myocardial ischemia: overview and outlook. Free Radic Biol Med 1988; 4: 9–14, doi: 10.1016/0891-5849(88)90005-6.
https://doi.org/10.1016/0891-5849(88)900...
). These ROS can initiate chain reactions in tissues, leading to irreversible damage in proteins, lipids, and nucleic acids (11. Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med 2000; 109: 315–323, doi: 10.1016/S0002-9343(00)00467-8.
https://doi.org/10.1016/S0002-9343(00)00...
). ROS play an essential role in regulating cell activities, such as gene expression, cell growth, and cell death (33. Radomska-Lesniewska DM, Hevelke A, Skopinski P, Balan B, Jozwiak J, Rokicki D, et al. Reactive oxygen species and synthetic antioxidants as angiogenesis modulators: clinical implications. Pharmacol Rep 2016; 68: 462–471, doi: 10.1016/j.pharep.2015.10.002.
https://doi.org/10.1016/j.pharep.2015.10...
). ROS can be detoxified by endogenous enzymes or free radical scavengers. However, the imbalance between antioxidants and oxidants leads to overproduction of ROS. This effect is associated with many multifactorial diseases, such as cardiovascular disorders (44. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the american heart association. Circ Res 2016; 119: e39-e75, doi: 10.1161/RES.0000000000000110.
https://doi.org/10.1161/RES.000000000000...
,55. Chen K, Keaney JF Jr. Evolving concepts of oxidative stress and reactive oxygen species in cardiovascular disease. Curr Atheroscler Rep 2012; 14: 476–483, doi: 10.1007/s11883-012-0266-8.
https://doi.org/10.1007/s11883-012-0266-...
). Global ischemia and reperfusion have been associated with the upregulation of ROS in cardiomyocytes (e.g., hydrogen peroxide [H2O2]), resulting in oxidative stress injuries (66. Venardos KM, Perkins A, Headrick J, Kaye DM. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr Med Chem 2007; 14: 1539–1549, doi: 10.2174/092986707780831078.
https://doi.org/10.2174/0929867077808310...
). H2O2 may cause apoptosis in cardiomyocytes by activating the intrinsic apoptotic pathways. Therefore, H2O2 is often utilized to establish an in vitro model for ischemia and subsequent reperfusion (I/R) injury (44. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the american heart association. Circ Res 2016; 119: e39-e75, doi: 10.1161/RES.0000000000000110.
https://doi.org/10.1161/RES.000000000000...
,55. Chen K, Keaney JF Jr. Evolving concepts of oxidative stress and reactive oxygen species in cardiovascular disease. Curr Atheroscler Rep 2012; 14: 476–483, doi: 10.1007/s11883-012-0266-8.
https://doi.org/10.1007/s11883-012-0266-...
).
Hydrogen sulfide (H2S) is predominantly synthesized from L-cysteine via cystathionine γ-lyase in the heart and vasculature. H2S has drawn great scientific interest regarding myocardial protection. H2S possesses almost all the beneficial cardiovascular effects similar to another well-characterized gasotransmitter, nitric oxide (NO) (77. Martelli A, Testai L, Breschi MC, Blandizzi C, Virdis A, Taddei S, et al. Hydrogen sulphide: novel opportunity for drug discovery. Med Res Rev 2012; 32: 1093–1130, doi: 10.1002/med.20234.
https://doi.org/10.1002/med.20234...
). Moreover, H2S acts as a ROS scavenger without producing deleterious ROS, which is typically seen in NO. H2S is rapidly emerging as a novel lipophilic cytoprotective signaling molecule with potent antioxidant, anti-inflammatory, and anti-apoptotic features (88. Lefer DJ. A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc Natl Acad Sci USA 2007; 104: 17907–17908, doi: 10.1073/pnas.0709010104.
https://doi.org/10.1073/pnas.0709010104...
,99. Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res 2009; 105: 365–374, doi: 10.1161/CIRCRESAHA.109.199919.
https://doi.org/10.1161/CIRCRESAHA.109.1...
). Our previous in vivo study revealed that the exogenous H2S donor, sodium hydrosulfide (NaHS), has potent anti-inflammatory effects in the heart damaged by acute myocardial infarction, which may be partially due to the limited CD11b+Gr-1+ myeloid cells (1010. Zhang Y, Li H, Zhao G, Sun A, Zong NC, Li Z, et al. Hydrogen sulfide attenuates the recruitment of CD11b(+)Gr-1(+) myeloid cells and regulates Bax/Bcl-2 signaling in myocardial ischemia injury. Sci Rep 2014; 4: 4774, doi: 10.1038/srep04774.
https://doi.org/10.1038/srep04774...
).
Although we have discovered the physiological and cardioprotective effects of H2S, the signaling mechanisms that mediate these effects have not been thoroughly evaluated. This study aimed to elucidate the mechanisms by which H2S prevents apoptosis of cardiomyocytes. In particular, we studied the mitochondrial pathway in response to H2O2-induced oxidative stress using an in vitro model that mimicked ischemia-reperfusion injuries.
Material and Methods
H2S donor
H2S was administered in the form of sodium hydrosulfide (NaHS) (Sigma-Aldrich, Cat. No.161527, USA). NaHS was freshly prepared in normal saline (0.9%) to the desired concentration before administration.
Cell culture
H9c2 cells (rat cardiomyoblasts) were obtained from the Cell Bank of the Chinese Academy of Sciences (China), and grown at a density of 105 cells/cm2 as a monolayer. H9c2 cells were cultured in Dulbecco's modified Eagle medium (GIBCO, Cat. No. 11960-077, USA) supplemented with 10% v/v fetal bovine serum, 2 mM glutamine, 1% nonessential amino acids, 100 IU penicillin, and 100 μg/mL streptomycin under an atmosphere of 5% CO2 saturated with water vapor at 37°C. The medium was replaced by fresh medium every two days. Subculture was done when the plates were more than 90% confluency.
Hydrogen peroxide treatment
To induce oxidative stress in H9c2 cells, the cells were cultured in serum-free medium containing 100 μM H2O2 for 24 h with or without a 30 min pre-treatment of 100 μM NaHS. H2O2 and NaHS were freshly prepared before each experiment. Control groups were treated with both H2O2 and NaHS simultaneously.
Apoptosis assay
An annexin V apoptosis detection kit (BD Biosciences, Cat. No. 556547, USA) was utilized to measure apoptosis of H2c9 cells following the manufacturer's instruction. After treatments, cells were washed twice with cold PBS, trypsinized, and then resuspended in the binding buffer at a concentration of 1×106 cells/mL. A 100 μL-aliquot of the cell suspension (1×105 cells) was then incubated with fluorescein isothiocyanate (FITC)-annexin V and propidium iodide for 15 min at room temperature in the dark. Apoptotic rate was analyzed using flow cytometry within 1 h.
Measurement of ROS production
The presence of intracellular ROS was detected using dihydroethidium (DHE, Sigma, Cat. No. D7008, USA), a sensitive fluorescent dye. According to the manufacturer's instructions, sub-confluent cells were pre-treated with or without 100 μM NaHS for 30 min, and then further incubated with 100 μM H2O2 for 24 h. Cells were then washed with PBS and incubated with 5 μM DHE at 37°C for 30 min. Fluorescence was captured with a fluorescent microscope, and the signal intensity was reported as a percentage of the control group.
Measurement of mitochondrial membrane potential (&b_psgr;m)
Changes in the ψm were detected using a mitochondria-specific cationic dye, JC-1 (Life Technologies, Cat. No. T3168, USA). JC-1 is a lipophilic cation that can selectively enter into mitochondria. H9c2 cells with or without H2O2 treatment were incubated with 10 μg/mL JC-1 at 37°C in the dark for 10 min. The loading solution was then replaced with fresh medium, and the fluorescent signal was captured and analyzed using the fluorescence microscopy. Red emission indicates membrane potential-dependent JC-1 aggregates in mitochondria. Green fluorescence reflects the monomeric form of JC-1 appearing in the cytoplasm after mitochondrial membrane depolarization. Consequently, mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio.
Western blot analysis
Western blot analysis was performed as previously described (1111. Zhang Y, Wang J, Li H, Yuan L, Wang L, Wu B, et al. Hydrogen sulfide suppresses transforming growth factor-beta1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Sci China Life Sci 2015; 58: 1126–1134, doi: 10.1007/s11427-015-4904-6.
https://doi.org/10.1007/s11427-015-4904-...
). Total protein lysates were collected for the standard immunoblot analysis. Protein concentrations were determined by a BCA protein assay. Aliquots of protein lysates (30 μg/lane) were loaded into sodium dodecyl sulfide-polyacrylamide gels (SDS-PAGE) and transferred to the PVDF membrane. The membrane was blocked and incubated with Bcl-2, Bax, and activated-caspase 3 p17 primary antibodies overnight at 4°C. Bcl-2 and Bax antibodies were purchased from Cell Signaling Technology (USA) (Bcl-2 Cat. No. 3498; Bax Cat. No. 14796), and activated-caspase 3 p17 antibody was from Bioworld (Cat. No. BS7004, USA). Blots were washed with TBST (Sigma-Aldrich), followed by incubation with corresponding horseradish peroxidase-conjugated secondary antibodies (Jackson Laboratory, USA). Lastly, the blots were visualized with enhanced chemiluminescence and were quantified by densitometry.
Statistical analysis
Data are reported as means±SE. Different groups were compared using one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls post hoc test. Comparison between two groups was assessed by t-test. P<0.05 was considered statistically significant.
Results
H2S reduced H2O2-induced apoptosis
To determine whether H2S affected H2O2-induced apoptosis in H2c9 cells, we performed annexin V & PI assays. Flow cytometry was then utilized to evaluate the apoptotic rate (Figure 1A). Compared to the control group, H2O2 significantly induced cell apoptosis, but the presence of H2S rescued H2O2 cytotoxicity, and increased cell viability (Figure 1B).
Hydrogen sulfide (H2S) reduced H2O2-induced H9c2 apoptosis. A, Cell death analysis of treated cells was performed by flow cytometry with annexin V/PI double staining. Representative quantitative analysis is shown in B. Data are reported as means±SE (n=6). *P<0.05, **P<0.01 vs control; #P<0.05, ##P<0.01 vs NaHS; &p<0.01 vs H2O2 (ANOVA followed by Student-Newman-Keuls post hoc test).
H2S protected H9c2 cells from oxidative stress
Oxidative stress leads to detrimental changes in the cell signaling process. To determine whether H2S has any effect on ROS activity under oxidative stress, we examined total ROS levels using DHE. Compared to the control group, H9c2 cells with H2O2 treatment showed an enhancement of fluorescence intensity by approximately six-fold. However, pre-treatment with H2S demonstrated a significant rescue effect (50.2% reduction of the fluorescence intensity) (Figure 2).
Hydrogen sulfide (H2S) protected H9c2 cells against H2O2-induced oxidative stress (A). Intracellular superoxide anion production was detected with dihydroethidium and observed by fluorescent microscopy (B). The fluorescent signal was measured and quantified. Data are reported as means±SE (n=6). *P<0.01 vs control; #P<0.01 vs NaHS; &P<0.01 vs H2O2 (ANOVA followed by Student-Newman-Keuls post hoc test). ROS: reactive oxygen species.
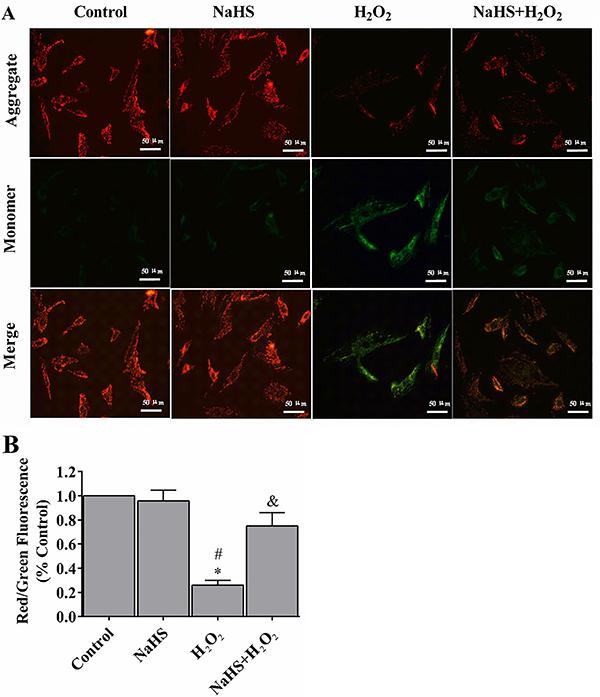
H2S prevented the loss of mitochondrial membrane potential (&b_psgr;m)
Mitochondrial function is highly sensitive to oxidative damages. In this study, we applied the fluorescent dye JC-1 to investigate whether H2S protected the physical functions of mitochondria from H2O2-induced cell stress in the cell model. In the control group, H9c2 cells exhibited numerous brightly stained mitochondria that emitted orange fluorescence (Figure 3A). After treating with H2O2 for 24 h, H9c2 cells demonstrated fewer and less stained mitochondria (Figure 3B). H2S pre-treatment, on the other hand, prevented H2O2-induced loss of mitochondrial membrane potential.
Hydrogen sulfide (H2S) prevented the loss of mitochondrial membrane potential (ψm) (A). The ψm loss was determined by the lipophilic cationic probe JC-1. Red signal indicates JC-1 in mitochondria and green signal indicates cytosolic JC-1. Magnification ×400; bar: 50 μm. B, Quantitative analysis of membrane potential. Data are reported as means±SE (n=6). *P<0.01 vs control; #P<0.01 vs NaHS; &P<0.05 vs H2O2 (ANOVA followed by the Student-Newman-Keuls post hoc test).
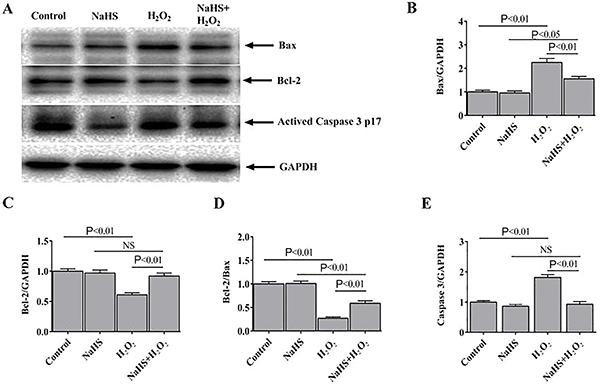
H2S regulated the expression of apoptosis-related proteins
Compared to the expression levels of H2O2-treated H9c2 cells, the protein expression level of Bax was significantly reduced, while Bcl-2 expression was increased in the NaHS-treated cells (Figure 4A). Consequently, compared with the H2O2 group, the ratio of Bcl-2 to Bax was higher in the NaHS + H2O2 group (Figure 4B-D). The expression level of activated caspase 3, another apoptotic marker, was also examined (Figure 4E). The results suggested that H2O2 enhanced apoptosis in H9c2 cells, while the NaHS treatments decreased H2O2-induced apoptosis.
Hydrogen sulfide (H2S) regulated the expression of apoptosis-related proteins. A, Representative immunoblots showing the expression of Bax, Bcl-2, and activated caspase 3 p17 in the H9c2 cells. Bax (B) and Bcl-2 (C) expression normalized to GAPDH (n=6). D, Densitometric analysis of the ratio of Bcl-2 to Bax. E, Activated caspase 3 p17 expression normalized to GAPDH. Data are reported as means±SE (n=6) (ANOVA followed by Student-Newman-Keuls post hoc test). NS: not significant.
Discussion
As a gaseous signaling molecule, H2S can freely diffuse across cell membranes and activate various cellular targets. This distinct feature makes H2S an attractive pharmacological agent to treat cardiovascular diseases. Previous studies have demonstrated a compelling cardioprotective effect of H2S in rat and mouse models (1212. Xie X, Sun A, Zhu W, Huang Z, Hu X, Jia J, et al. Transplantation of mesenchymal stem cells preconditioned with hydrogen sulfide enhances repair of myocardial infarction in rats. Tohoku J Exp Med 2012; 226: 29–36, doi: 10.1620/tjem.226.29.
https://doi.org/10.1620/tjem.226.29...
13. Liu Z, Han Y, Li L, Lu H, Meng G, Li X, et al. Hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E mice. British J Pharmacol 2013; 169: 1795–1809, doi: 10.1111/bph.12246.
https://doi.org/10.1111/bph.12246...
–1414. Ma SF, Luo Y, Ding YJ, Chen Y, Pu SX, Wu HJ, et al. Hydrogen sulfide targets the cys320/cys529 motif in kv4.2 to inhibit the ito potassium channels in cardiomyocytes and regularizes fatal arrhythmia in myocardial infarction. Antioxid Redox Signal 2015; 23: 129–147, doi: 10.1089/ars.2014.6094.
https://doi.org/10.1089/ars.2014.6094...
). We have shown that H2S pre-treatment efficiently protected human ventricular fibroblasts from H2O2-induced endoplasmic reticulum (ER) stress and prevented the activation of caspase cascade (1515. Feng A, Ling C, Xin Duo L, Bing W, San Wu W, Yu Z, et al. Hydrogen sulfide protects human cardiac fibroblasts against H2O2-induced injury through regulating autophagy-related proteins. Cell Transplant 2018; 27: 1222–1234, doi: 10.1177/0963689718779361.
https://doi.org/10.1177/0963689718779361...
). In the current study, we showed that H2S protected H9c2 cardioblasts from H2O2-induced oxidative stress. H2S regulated the cell cycle, decreased apoptotic cells, and preserved mitochondrial membrane potential ψm that is essential for ATP production and homeostasis. H2S also upregulated Bcl-2/Bax ratio, suggesting its critical role in the anti-apoptotic mechanisms. Both studies indicated the holistic role of H2S in protecting the cardiovascular system.
Oxidative stress is the imbalance between ROS production and detoxification. This imbalance impairs the capacity to repair the damages caused by reactive intermediates. Moreover, imbalance in oxidative states may lead to the generation of peroxides and free radicals, which in turn damage proteins, lipids, and DNA (11. Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med 2000; 109: 315–323, doi: 10.1016/S0002-9343(00)00467-8.
https://doi.org/10.1016/S0002-9343(00)00...
). Oxidative damage from H2O2 contributes to heart failure and tissue damages (1616. Seo YJ, Lee JW, Lee EH, Lee HK, Kim HW, Kim YH. Role of glutathione in the adaptive tolerance to H2O2. Free Rad Biol Med 2004; 37: 1272–1281, doi: 10.1016/j.freeradbiomed.2004.07.012.
https://doi.org/10.1016/j.freeradbiomed....
). The H2O2 molecule plays an essential role in the progression of oxidative stress and cardiac pathologies (1717. Slezak J, Tribulova N, Pristacova J, Uhrik B, Thomas T, Khaper N, et al. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and X-ray microanalysis. Am J Pathol 1995; 147: 772–781.,1818. Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res 2011; 108: 566–573, doi: 10.1161/CIRCRESAHA.110.237636.
https://doi.org/10.1161/CIRCRESAHA.110.2...
). In addition, excessive ROS damages mitochondria, opens its permeability transition pore (PTP), and thus induces mitochondrial permeability transition. These alterations cause mitochondrial depolarization and outer membrane rupture, leading to cell apoptosis or death (1919. Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007; 12: 815–833, doi: 10.1007/s10495-007-0723-y.
https://doi.org/10.1007/s10495-007-0723-...
,2020. Rannikko EH, Vesterager LB, Shaik JH, Weber SS, Cornejo Castro EM, Fog K, et al. Loss of DJ-1 protein stability and cytoprotective function by Parkinson's disease-associated proline-158 deletion. J Neurochem 2013; 125: 314–327, doi: 10.1111/jnc.12126.
https://doi.org/10.1111/jnc.12126...
). Apoptosis that occurs in the clinical setting (e.g., open-heart surgery under cardiopulmonary bypass) is induced by various conditions and agents, including ROS, NO, calcium, and pressure overload, mechanical stress, tumor necrosis factor, and angiotensin II. Apoptosis has also been shown to play a pivotal role in the pathogenesis of ischemia/reperfusion (2121. Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am J Physiol Cell Physiol 2008; 294: C833-C841, doi: 10.1152/ajpcell.00313.2007.
https://doi.org/10.1152/ajpcell.00313.20...
).
Endogenous gaseous signaling mediators, such as H2S, are formed in mammalian cells and tissues. H2S may easily react with certain compounds, especially with reactive oxygen and nitrogen species, such as superoxide radical anion (O2-), H2O2, peroxynitrite (ONOO-), and hypochlorite (ClO-) (2222. Mitsuhashi H, Yamashita S, Ikeuchi H, Kuroiwa T, Kaneko Y, Hiromura K, et al. Oxidative stress-dependent conversion of hydrogen sulfide to sulfite by activated neutrophils. Shock 2005; 24: 529–534, doi: 10.1097/01.shk.0000183393.83272.de.
https://doi.org/10.1097/01.shk.000018339...
23. Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem 2004; 90: 765–768, doi: 10.1111/j.1471-4159.2004.02617.x.
https://doi.org/10.1111/j.1471-4159.2004...
–2424. Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, et al. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem Biophys Res Commun 2005; 326: 794–798, doi: 10.1016/j.bbrc.2004.11.110.
https://doi.org/10.1016/j.bbrc.2004.11.1...
). In the current study, we demonstrated that H2O2 induced ROS production and decreased mitochondrial membrane potential, suggesting an impairment of mitochondrial functions. H2S treatment, however, attenuated the ROS production, restored mitochondrial membrane potential, and decreased cardiomyocyte apoptosis. H2S may protect mitochondrial function via multiple pathways, such as activating AMPK in cardiomyocytes (2525. Wagner F, Asfar P, Calzia E, Radermacher P, Szabo C. Bench-to-bedside review: Hydrogen sulfide--the third gaseous transmitter: applications for critical care. Crit Care 2009; 13: 213, doi: 10.1186/cc7700.
https://doi.org/10.1186/cc7700...
).
Previous studies showed that Bcl-2 family is upregulated during the opening of PTP (1919. Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007; 12: 815–833, doi: 10.1007/s10495-007-0723-y.
https://doi.org/10.1007/s10495-007-0723-...
,2020. Rannikko EH, Vesterager LB, Shaik JH, Weber SS, Cornejo Castro EM, Fog K, et al. Loss of DJ-1 protein stability and cytoprotective function by Parkinson's disease-associated proline-158 deletion. J Neurochem 2013; 125: 314–327, doi: 10.1111/jnc.12126.
https://doi.org/10.1111/jnc.12126...
). PEP-1-CAT, a fusion protein of anti-microbial peptide and antioxidant enzyme, protects cardiomyocyte from hypoxia/reoxygenation-induced injuries. PEP-1-CAT blocks Bcl-2/Bax/mitochondrial apoptotic pathway by inhibiting p38 MAPK while activating the PI3K/Akt and Erk1/2 signaling pathways (2626. Zhang L, Wei S, Tang JM, Guo LY, Zheng F, Yang JY, et al. PEP-1-CAT protects hypoxia/reoxygenation-induced cardiomyocyte apoptosis through multiple sigaling pathways. J Transl Med 2013; 11: 113, doi: 10.1186/1479-5876-11-113.
https://doi.org/10.1186/1479-5876-11-113...
). Here, we also found H2S regulated the Bcl-2/Bax/caspase-3 signaling pathway in the rat cardiomyoblasts. The functions of Bcl-2 (anti-apoptosis) and Bax (pro-apoptosis) proteins are in opposition of the apoptotic pathway. When the apoptotic signal is present, caspase 3 (32 kD) will be cleaved and present as an active form (17 kD) to induce cell apoptosis (2727. Wang X, Wang Q, Guo W, Zhu YZ. Hydrogen sulfide attenuates cardiac dysfunction in a rat model of heart failure: a mechanism through cardiac mitochondrial protection. Biosci Rep 2011; 31: 87–98, doi: 10.1042/BSR20100003.
https://doi.org/10.1042/BSR20100003...
). In our study, H2O2 treatment led to a shift in favor of the pro-apoptotic protein, Bax, as well as upregulated the downstream effector, caspase 3. On the other hand, H2S rescued this apoptotic event and downregulated the level of caspase 3, demonstrating a cytoprotective effect in the rat cardiomyoblasts.
In conclusion, we demonstrated that NaHS has potent anti-apoptotic effects in cardiomyoblasts with H2O2-induced injuries. The anti-apoptotic function may be partially due to blocking ROS production in mitochondria, maintaining mitochondrial membrane integrity, and inhibiting activation of the Bcl-2/Bax apoptotic pathway. The current study suggested that H2S may serve as an effective therapeutic option for treating ischemia-reperfusion injuries.
Acknowledgments
This study was supported by the National Nature Science Foundation of China (No. 81500237 and 81701891) and Special Foundation for Knowledge Innovation of Hubei Province (Nature Science Foundation) (No. 2017CFB563).
References
-
1Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med 2000; 109: 315–323, doi: 10.1016/S0002-9343(00)00467-8.
» https://doi.org/10.1016/S0002-9343(00)00467-8 -
2McCord JM. Free radicals and myocardial ischemia: overview and outlook. Free Radic Biol Med 1988; 4: 9–14, doi: 10.1016/0891-5849(88)90005-6.
» https://doi.org/10.1016/0891-5849(88)90005-6 -
3Radomska-Lesniewska DM, Hevelke A, Skopinski P, Balan B, Jozwiak J, Rokicki D, et al. Reactive oxygen species and synthetic antioxidants as angiogenesis modulators: clinical implications. Pharmacol Rep 2016; 68: 462–471, doi: 10.1016/j.pharep.2015.10.002.
» https://doi.org/10.1016/j.pharep.2015.10.002 -
4Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the american heart association. Circ Res 2016; 119: e39-e75, doi: 10.1161/RES.0000000000000110.
» https://doi.org/10.1161/RES.0000000000000110 -
5Chen K, Keaney JF Jr. Evolving concepts of oxidative stress and reactive oxygen species in cardiovascular disease. Curr Atheroscler Rep 2012; 14: 476–483, doi: 10.1007/s11883-012-0266-8.
» https://doi.org/10.1007/s11883-012-0266-8 -
6Venardos KM, Perkins A, Headrick J, Kaye DM. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr Med Chem 2007; 14: 1539–1549, doi: 10.2174/092986707780831078.
» https://doi.org/10.2174/092986707780831078 -
7Martelli A, Testai L, Breschi MC, Blandizzi C, Virdis A, Taddei S, et al. Hydrogen sulphide: novel opportunity for drug discovery. Med Res Rev 2012; 32: 1093–1130, doi: 10.1002/med.20234.
» https://doi.org/10.1002/med.20234 -
8Lefer DJ. A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc Natl Acad Sci USA 2007; 104: 17907–17908, doi: 10.1073/pnas.0709010104.
» https://doi.org/10.1073/pnas.0709010104 -
9Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res 2009; 105: 365–374, doi: 10.1161/CIRCRESAHA.109.199919.
» https://doi.org/10.1161/CIRCRESAHA.109.199919 -
10Zhang Y, Li H, Zhao G, Sun A, Zong NC, Li Z, et al. Hydrogen sulfide attenuates the recruitment of CD11b(+)Gr-1(+) myeloid cells and regulates Bax/Bcl-2 signaling in myocardial ischemia injury. Sci Rep 2014; 4: 4774, doi: 10.1038/srep04774.
» https://doi.org/10.1038/srep04774 -
11Zhang Y, Wang J, Li H, Yuan L, Wang L, Wu B, et al. Hydrogen sulfide suppresses transforming growth factor-beta1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Sci China Life Sci 2015; 58: 1126–1134, doi: 10.1007/s11427-015-4904-6.
» https://doi.org/10.1007/s11427-015-4904-6 -
12Xie X, Sun A, Zhu W, Huang Z, Hu X, Jia J, et al. Transplantation of mesenchymal stem cells preconditioned with hydrogen sulfide enhances repair of myocardial infarction in rats. Tohoku J Exp Med 2012; 226: 29–36, doi: 10.1620/tjem.226.29.
» https://doi.org/10.1620/tjem.226.29 -
13Liu Z, Han Y, Li L, Lu H, Meng G, Li X, et al. Hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E mice. British J Pharmacol 2013; 169: 1795–1809, doi: 10.1111/bph.12246.
» https://doi.org/10.1111/bph.12246 -
14Ma SF, Luo Y, Ding YJ, Chen Y, Pu SX, Wu HJ, et al. Hydrogen sulfide targets the cys320/cys529 motif in kv4.2 to inhibit the ito potassium channels in cardiomyocytes and regularizes fatal arrhythmia in myocardial infarction. Antioxid Redox Signal 2015; 23: 129–147, doi: 10.1089/ars.2014.6094.
» https://doi.org/10.1089/ars.2014.6094 -
15Feng A, Ling C, Xin Duo L, Bing W, San Wu W, Yu Z, et al. Hydrogen sulfide protects human cardiac fibroblasts against H2O2-induced injury through regulating autophagy-related proteins. Cell Transplant 2018; 27: 1222–1234, doi: 10.1177/0963689718779361.
» https://doi.org/10.1177/0963689718779361 -
16Seo YJ, Lee JW, Lee EH, Lee HK, Kim HW, Kim YH. Role of glutathione in the adaptive tolerance to H2O2. Free Rad Biol Med 2004; 37: 1272–1281, doi: 10.1016/j.freeradbiomed.2004.07.012.
» https://doi.org/10.1016/j.freeradbiomed.2004.07.012 -
17Slezak J, Tribulova N, Pristacova J, Uhrik B, Thomas T, Khaper N, et al. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and X-ray microanalysis. Am J Pathol 1995; 147: 772–781.
-
18Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res 2011; 108: 566–573, doi: 10.1161/CIRCRESAHA.110.237636.
» https://doi.org/10.1161/CIRCRESAHA.110.237636 -
19Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007; 12: 815–833, doi: 10.1007/s10495-007-0723-y.
» https://doi.org/10.1007/s10495-007-0723-y -
20Rannikko EH, Vesterager LB, Shaik JH, Weber SS, Cornejo Castro EM, Fog K, et al. Loss of DJ-1 protein stability and cytoprotective function by Parkinson's disease-associated proline-158 deletion. J Neurochem 2013; 125: 314–327, doi: 10.1111/jnc.12126.
» https://doi.org/10.1111/jnc.12126 -
21Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am J Physiol Cell Physiol 2008; 294: C833-C841, doi: 10.1152/ajpcell.00313.2007.
» https://doi.org/10.1152/ajpcell.00313.2007 -
22Mitsuhashi H, Yamashita S, Ikeuchi H, Kuroiwa T, Kaneko Y, Hiromura K, et al. Oxidative stress-dependent conversion of hydrogen sulfide to sulfite by activated neutrophils. Shock 2005; 24: 529–534, doi: 10.1097/01.shk.0000183393.83272.de.
» https://doi.org/10.1097/01.shk.0000183393.83272.de -
23Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem 2004; 90: 765–768, doi: 10.1111/j.1471-4159.2004.02617.x.
» https://doi.org/10.1111/j.1471-4159.2004.02617.x -
24Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, et al. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem Biophys Res Commun 2005; 326: 794–798, doi: 10.1016/j.bbrc.2004.11.110.
» https://doi.org/10.1016/j.bbrc.2004.11.110 -
25Wagner F, Asfar P, Calzia E, Radermacher P, Szabo C. Bench-to-bedside review: Hydrogen sulfide--the third gaseous transmitter: applications for critical care. Crit Care 2009; 13: 213, doi: 10.1186/cc7700.
» https://doi.org/10.1186/cc7700 -
26Zhang L, Wei S, Tang JM, Guo LY, Zheng F, Yang JY, et al. PEP-1-CAT protects hypoxia/reoxygenation-induced cardiomyocyte apoptosis through multiple sigaling pathways. J Transl Med 2013; 11: 113, doi: 10.1186/1479-5876-11-113.
» https://doi.org/10.1186/1479-5876-11-113 -
27Wang X, Wang Q, Guo W, Zhu YZ. Hydrogen sulfide attenuates cardiac dysfunction in a rat model of heart failure: a mechanism through cardiac mitochondrial protection. Biosci Rep 2011; 31: 87–98, doi: 10.1042/BSR20100003.
» https://doi.org/10.1042/BSR20100003
Publication Dates
-
Publication in this collection
15 Apr 2019 -
Date of issue
2019
History
-
Received
11 Sept 2018 -
Accepted
6 Dec 2018