Abstract
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are the predominant mediators of glutamate-induced excitatory neurotransmission. It is widely accepted that AMPA receptors are critical for the generation and spread of epileptic seizure activity. Dysfunction of AMPA receptors as a causal factor in patients with intractable epilepsy results in neurotransmission failure. Brain-specific serine/threonine-protein kinase 1 (SAD-B), a serine-threonine kinase specifically expressed in the brain, has been shown to regulate AMPA receptor-mediated neurotransmission through a presynaptic mechanism. In cultured rat hippocampal neurons, the overexpression of SAD-B significantly increases the frequency of miniature excitatory postsynaptic currents (mEPSCs). Here, we showed that SAD-B downregulation exerted antiepileptic activity by regulating AMPA receptors in patients with temporal lobe epilepsy (TLE) and in the pentylenetetrazol (PTZ)-induced epileptic model. We first used immunoblotting and immunohistochemistry analysis to demonstrate that SAD-B expression was increased in the epileptic rat brain. Subsequently, to explore the function of SAD-B in epilepsy, we used siRNA to knock down SAD-B protein and observed behavior after PTZ-induced seizures. We found that SAD-B downregulation attenuated seizure severity and susceptibility in the PTZ-induced epileptic model. Furthermore, we showed that the antiepileptic effect of SAD-B downregulation on PTZ-induced seizure was abolished by CNQX (an AMPA receptor inhibitor), suggesting that SAD-B modulated epileptic seizure by regulating AMPA receptors in the brain. Taken together, these findings suggest that SAD-B may be a potential and novel therapeutic target to limit epileptic seizures.
SAD-B; AMPA receptor; Temporal lobe epilepsy; PTZ
Introduction
Epilepsy is a common chronic neurological disease characterized by recurrent spontaneous seizures. It is refractory to current therapies in approximately 30% of patients and is associated with pathologic cortical excitability (11. Dixit AB, Tripathi M, Chandra PS, Banerjee J. Molecular biomarkers in drug-resistant epilepsy: facts & possibilities. Int J Surg 2016; 36: 483–491, doi: 10.1016/j.ijsu.2015.08.029.
https://doi.org/10.1016/j.ijsu.2015.08.0...
). Temporal lobe epilepsy (TLE), the most prevalent form of refractory epilepsy, representing more than 40% of cases, is intractable (22. Blümcke I, Aronica E, Miyata H, Sarnat HB, Thom M, Roessler K, et al. International recommendation for a comprehensive neuropathologic workup of epilepsy surgery brain tissue: a consensus Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 2016; 57: 348–358, doi: 10.1111/epi.13319.
https://doi.org/10.1111/epi.13319...
) and results in serious cognitive impairment, sudden unexpected death in epilepsy (SUDEP), vascular disease, pneumonia, etc. (33. Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav 2014; 37: 59–70, doi: 10.1016/j.yebeh.2014.05.031.
https://doi.org/10.1016/j.yebeh.2014.05....
). In terms of the pathogenesis of epilepsy, it is widely accepted that an imbalance in neuronal excitation and inhibition results in neurotransmission failure as a causal factor in patients with TLE. α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, located at the postsynaptic membrane of excitatory synapses, are the predominant mediators of glutamate-induced excitatory neurotransmission in the neural system (44. Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr 2011; 11: 56–63, doi: 10.5698/1535-7511-11.2.56.
https://doi.org/10.5698/1535-7511-11.2.5...
). Dysfunction of AMPA receptors as one of the most important factors in patients with epilepsy has been extensively reviewed (55. Di Bonaventura C, Labate A, Maschio M, Meletti S, Russo E. AMPA receptors and perampanel behind selected epilepsies: current evidence and future perspectives. Expert Opin Pharmacother 2017; 18: 1751–1764, doi: 10.1080/14656566.2017.1392509.
https://doi.org/10.1080/14656566.2017.13...
). Thus, there has been considerable interest in developing therapeutic strategies aimed at AMPA receptors.
Brain-specific serine/threonine-protein kinase 1 (BRSK1, also referred to as SAD-B), a mammalian serine/threonine kinase, is specifically expressed in the brain (66. Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, et al. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 2007; 129: 549–563, doi: 10.1016/j.cell.2007.03.025.
https://doi.org/10.1016/j.cell.2007.03.0...
). SAD-B is involved in promoting the maturation of nerve terminals and modulating neurotransmitter release and regulates correct synaptic development and presynaptic vesicle clustering in both sensory and motor neurons (77. Crump JG, Zhen M, Jin Y, Bargmann CI. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron 2011; 29: 115–129, doi: 10.1016/S0896-6273(01)00184-2.
https://doi.org/10.1016/S0896-6273(01)00...
). Notably, SAD-B associates with synaptic vesicles at the active zone cytomatrix of presynaptic terminals, and the overexpression of SAD-B significantly increases the frequency of miniature excitatory postsynaptic currents (mEPSCs) (88. Inoue E, Mochida S, Takagi H, Higa S, Deguchi-Tawarada M, Takao-Rikitsu E, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron 2006; 50: 261–275, doi: 10.1016/j.neuron.2006.03.018.
https://doi.org/10.1016/j.neuron.2006.03...
). Therefore, we hypothesized that SAD-B may modulate epileptic seizures by regulating AMPA receptor function in patients with TLE and in the PTZ-induced epileptic model.
In this study, we first observed that SAD-B expression was increased in patients with TLE and in the PTZ-induced epileptic model. Subsequently, we found SAD-B downregulation attenuated seizure severity and susceptibility in the PTZ-induced epileptic model. Furthermore, we found that the effect of SAD-B downregulation on PTZ-induced seizure was abolished by CNQX (an AMPA receptor inhibitor) (99. Kim M, Campeau S, Falls WA, Davis M. Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav Neural Biol 1993; 59: 5–8, doi: 10.1016/0163-1047(93)91075-X.
https://doi.org/10.1016/0163-1047(93)910...
), suggesting that the antiepileptic effect of SAD-B downregulation on PTZ-induced seizure was mediated by AMPA receptors. These results indicated that SAD-B may represent a potential therapeutic target for epilepsy.
Material and Methods
Human samples
Samples from twenty patients with TLE (12 males and 8 females; age: 23.35±9.05 years; epilepsy course: 8.35±4.68 years) were obtained from the brain tissue bank of First Affiliated Hospital of Chongqing Medical University. Pre-surgical assessments were thoroughly evaluated to ensure that the patients with epilepsy were suitable for surgery. Twenty histologically normal samples were obtained from patients who experienced head trauma (11 males and 9 females; age: 23.95±8.45 years), did not present with signs of apparent nervous system disease, and were not exposed to antiepileptic drugs (AEDs). The clinical features of the patients included in this study are reported in Tables 1 and 2. No significant difference in sex or age was observed between the two groups (P>0.05). The study protocols were performed according to the guidelines for the implementation of research on humans as established by the National Institutes of Health of China and the Committee on Human Research at Chongqing Medical University. Written informed consent for the use of human brain tissues was collected as described in our previous publication (1010. Yu X, Xu T, Ou S, Yuan J, Deng J, Li R, et al. Endophilin A1 mediates seizure activity via regulation of AMPARs in a PTZ-kindled epileptic mouse model. Exp Neurol 2018; 304: 41–57, doi: 10.1016/j.expneurol.2018.02.014.
https://doi.org/10.1016/j.expneurol.2018...
).
Experimental animals
Sprague-Dawley rats were obtained from the Laboratory Animal Center of Chongqing Medical University (China). In the chronic model, we administered a sub-convulsive dose of PTZ (35 mg/kg, ip, Sigma, USA) daily. The animals were considered to be kindled after exhibiting at least three consecutive stage 4 or 5 seizures (Racine's scale evaluation). In the acute PTZ-induced seizure model, we injected rats with PTZ (70 mg/kg, ip) to induce seizures. Diazepam (10 mg/kg) was administered by ip injection 1 h after the initial onset of seizures to stop the continuous seizures. All protocols for the handling and treatment of animals were approved by the Commission of Chongqing Medical University for Ethics in Animal Experiments (approval no. 0002648). All experiments were conducted according to the principles of Animal Research: Reporting in vivo Experiments (ARRIVE) guidelines (https://www.nc3rs.org.uk/arrive-guidelines). All efforts were made to decrease the number of animals and alleviate their suffering.
Tissue preparation
All human brain tissues were collected in the operating room as described in our previous publication (1010. Yu X, Xu T, Ou S, Yuan J, Deng J, Li R, et al. Endophilin A1 mediates seizure activity via regulation of AMPARs in a PTZ-kindled epileptic mouse model. Exp Neurol 2018; 304: 41–57, doi: 10.1016/j.expneurol.2018.02.014.
https://doi.org/10.1016/j.expneurol.2018...
). Minor modifications included the sectioning of the tissue into 30-µm thick sections for double immunofluorescence labelling.
Immunohistochemistry
Avidin-biotin-peroxidase complex (Vectastain Elite ABC; Vector Laboratories International, USA) was used for immunohistochemistry as described in our previous study (1010. Yu X, Xu T, Ou S, Yuan J, Deng J, Li R, et al. Endophilin A1 mediates seizure activity via regulation of AMPARs in a PTZ-kindled epileptic mouse model. Exp Neurol 2018; 304: 41–57, doi: 10.1016/j.expneurol.2018.02.014.
https://doi.org/10.1016/j.expneurol.2018...
). Minor modifications included the use of a polyclonal mouse SAD-B antibody (diluted 1:100, Cat. No. ab206298; Abcam, UK) as the primary antibody.
Double immunofluorescence labelling
Double immunofluorescence staining was conducted on five randomly selected sections as described previously (1010. Yu X, Xu T, Ou S, Yuan J, Deng J, Li R, et al. Endophilin A1 mediates seizure activity via regulation of AMPARs in a PTZ-kindled epileptic mouse model. Exp Neurol 2018; 304: 41–57, doi: 10.1016/j.expneurol.2018.02.014.
https://doi.org/10.1016/j.expneurol.2018...
). The primary antibodies included mouse anti-SAD-B (diluted 1:100, Cat. No. ab206298; Abcam, UK), chicken anti-microtubule-associated protein 2 (MAP2) (1:200, Cat. No. ab5392; Abcam), and rabbit anti-glial fibrillary acidic protein (GFAP) (1:50, Cat. No. BM0055; Boster, China). The secondary antibodies included Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:100; Proteintech, China), DyLight 405-conjugated goat anti-chicken IgG (1:50, Beyotime Institute of Biotechnology, China), and Alexa Fluor 549-conjugated goat anti-mouse IgG (1:100; Proteintech, China).
Immunoblotting
Human brain tissues from patients with TLE or traumatic brain injury, as well as rat cortices and hippocampi, were homogenized in RIPA lysis buffer (Proteintech, China). The supernatants were removed after centrifugation at 4°C (12,000 g, 15 min). Prepared proteins (40 μg per lane) were separated by SDS-polyacrylamide gel electrophoresis (SDS–PAGE; 5% spacer gel, 80 V, 30 min; 10% separating gel, 120 V, 90 min) before being transferred to a polyvinylidene fluoride (PVDF) membrane (220 mA, between 60 and 120 min according to the molecular weight of the protein). Next, the PVDF membrane was incubated at 37°C for 1 h in 10% skim milk in Tween-20 for blocking. The PVDF membrane was then incubated with polyclonal mouse anti-SAD-B-1 (diluted 1:100, Cat. No. ab206298; Abcam) diluted in 10% skim milk diluted in freshly prepared 5% BSA at 4°C overnight. The membrane was washed with Tween-20-Tris-buffered saline (TBS) for 45 min, with the solution changed every 10 min, and then incubated with goat anti-mouse IgG antibody or goat anti-rabbit IgG antibody (1:1000, Proteintech, China) for 1 h at 37°C. A rabbit anti-GAPDH antibody (1:1000, Proteintech) was used as the loading control. Densitometry quantitation was determined using Quantity One 1-D Analysis Software (Bio-Rad Laboratories, USA) as optical density values, and the SAD-B levels were normalized to those of GAPDH.
Co-immunoprecipitation
Human cortical and rat hippocampal tissues were homogenized and mixed with RIPA lysis buffer (Beyotime, China). Equal amounts of protein were then incubated with antibody (4 μL of polyclonal mouse anti-SAD-B1 (1:50 Cat. No. ab206298; Abcam, UK), 4 μL of mouse anti-GluA1–GluA4, (1:100, Santa Cruz Biotechnology, USA) or 2 μL of rabbit IgG, which served as a control (1:100, Abcam). The supernatant fractions were incubated for 12 h at 4°C and then incubated with 15 μL of protein A/G agarose beads (Beyotime) for 1.5 h at 4°C. The immunoprecipitates were washed twice with lysis buffer, boiled in 1× SDS loading buffer for 10 min and separated on SDS-polyacrylamide gels. Immunoblotting experiments were performed with anti-GluA1-GluA4 and anti-SAD-B antibodies.
Behavioral tests
Two observers who were blinded to the handling conditions were responsible for recording the general behaviors induced by PTZ. Seizures were induced between 8:00 and 10:00 am to minimize the possible complicating effects of the animals' circadian rhythms. For seizure severity, the behaviors were scored as follows (1111. Nutt DJ, Cowen PJ, Batts CC, Grahame-Smith DG, Green AR. Repeated administration of subconvulsant doses of GABA antagonist drugs. Psychopharmacology 1982; 76: 84–87, doi: 10.1007/BF00430762.
https://doi.org/10.1007/BF00430762...
): 0) no further seizures; 1) a generalized clonic seizure; 2) a generalized clonic seizure with loss of righting reflex; 3) a generalized clonic seizure with loss of righting reflex plus running and bouncing; and 4) all behaviors associated with a score of 3 plus forelimb tonus. Latency was recorded as the time after PTZ injection to the first seizure onset. The generalized tonic clonic (GTC) value per rat is the number of GTCs per rat within 10 min of PTZ injection.
Lentiviral vector generation and stereotaxic injection
Lentiviral (LV) vectors carrying siRNA were constructed by a ubiquitin promoter (GeneChem Co., Ltd., China). The control (no target siRNA) was the same lentiviral vector expressing GFP (LV-GFP). We performed microinjections of siRNA or LV-GFP into the bilateral CA1 region of the dorsal hippocampus of each animal in a total volume of 20 μL (10 μL per side) using a glass pipette (0.2 μL/min). Two weeks after the administration of the lentiviral vectors, we induced seizures with PTZ.
In vivo multichannel EEG recording and local field potential (LFP) analysis
After SAD-B-siRNA was injected into the CA1 region (AP –3.6 mm, ML –2.6 mm, and DV –2.8 mm), a microwire array (a 1.5×6 array of platinum-iridium alloy wires, each with a 20-μm diameter) was implanted into the same site in the CA1 region of the hippocampus. PTZ was administered to induce seizures (after two weeks). In vivo multichannel EEG recordings were continuously collected for 10 min after the administration of PTZ. According to the PTZ scoring criteria, an electrophysiological seizure was defined as the manifestation of seizure behaviors ranging from stages 1–4 in the rats. The intra-hippocampal LFP of electroencephalography (EEG) recordings showed a high-amplitude discharge (>3-fold higher the baseline) that began in the hippocampus and spread to the cortex with a high frequency (>5 Hz). Latencies and generalized tonic clonic seizures (GTCs) in the observed 10 min period during status epilepticus were recorded for each animal from each group. LFPs were preamplified (1000×), filtered (0.1–1000 Hz), and digitized at 4 kHz using an OmniPlex D Neural Data Acquisition System (Plexon, USA). All animals were euthanized with pentobarbital (100 mg/kg, ip, Sigma) after the seizures were recorded and hippocampal tissues were collected for morphological and biochemical studies.
Results
SAD-B was localized in the epileptic brain
Epilepsy is known to be characterized by neuronal hyperexcitability, thus, we first determined whether SAD-B is located in neurons. Immunofluorescence showed that SAD-B was localized in the cortical neurons of the patients with TLE (Figure 1A). In the PTZ-induced epileptic model, SAD-B was localized in the cortex and hippocampus of epileptic rats (Figure 1B). SAD-B was co-expressed with the neuron-specific marker MAP2 in neurons but not with the astrocyte-specific marker GFAP.

Brain-specific serine/threonine-protein kinase 1 (SAD-B) is localized in the epileptic brain. A, Immunofluorescence labelling of SAD-B (red), MAP2 (violet), and GFAP (green) in the cortex of patients with temporal lobe epilepsy (TLE) showing that SAD-B was co-localized with MAP2 but not with GFAP. Scale bar: 50 μm (400×). B, Immunofluorescence labelling of SAD-B (red), MAP2 (violet), and GFAP (green) in the CA1 region of the hippocampus or cortex of an epileptic rat showing that SAD-B was co-localized with MAP2, but not with GFAP. Scale bar: 50 μm (400×). White arrows: SAD-B; yellow arrows: GFAP.
SAD-B expression was increased in the epileptic brain
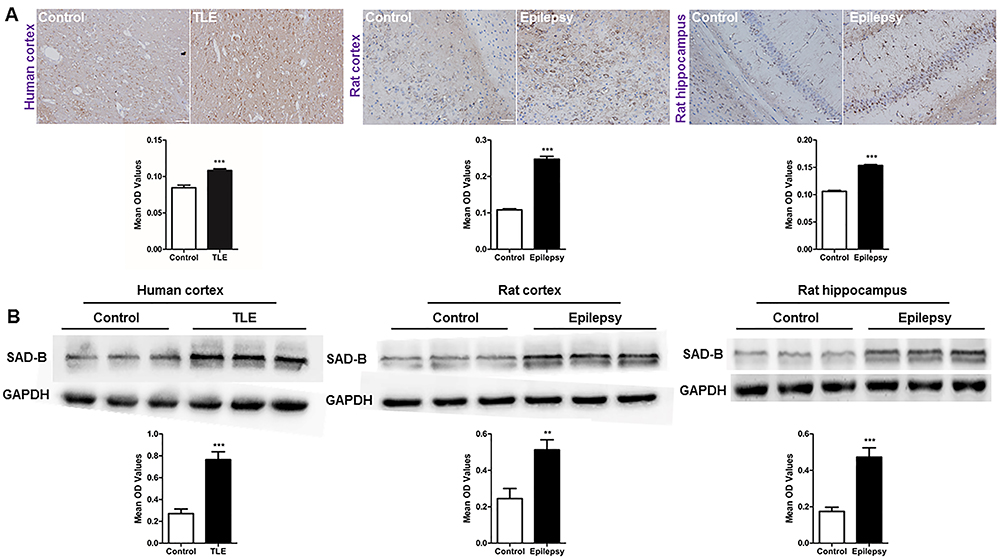
To examine whether SAD-B is involved in epileptic seizure, we examined SAD-B expression in the patients with TLE. Immunohistochemistry (IHC) showed that SAD-B expression was significantly increased in patients with TLE compared to the controls. Moreover, immunoblotting was performed and the results showed that SAD-B expression was also significantly increased in patients with TLE (Figure 2A). In epileptic rats, SAD-B expression was increased in the cortex and hippocampus compared to the controls (Figure 2B). These findings indicated that SAD-B may be involved in epileptic seizure in the brain.

Brain-specific serine/threonine-protein kinase 1 (SAD-B) expression is increased in the epileptic brain. A, Immunohistochemistry for SAD-B (left panels) showing that the average optical density (OD) of SAD-B in patients with temporal lobe epilepsy (TLE) was increased compared to that in the controls (n=5 pairs, ***P<0.05, unpaired t-test). In the epileptic rats, SAD-B expression was also increased in the cortices and hippocampi of PTZ-induced epileptic rats compared to that in the controls (n=5 pairs, ***P<0.05, unpaired t-test). Scale bar: 100 μm (400×). B, Immunoblot analysis showing that SAD-B expression was significantly increased in patients with TLE compared to controls (n=12 pairs, ***P<0.001, unpaired t-test). In the epileptic rats, SAD-B expression was also significantly increased in the cortices and hippocampi of the PTZ-induced rats compared to controls (n=12 pairs, **P<0.01, ***P<0.001, unpaired t-test). Data are reported as mean±SEM.
SAD-B downregulation reduced seizure severity and susceptibility in the PTZ-induced model
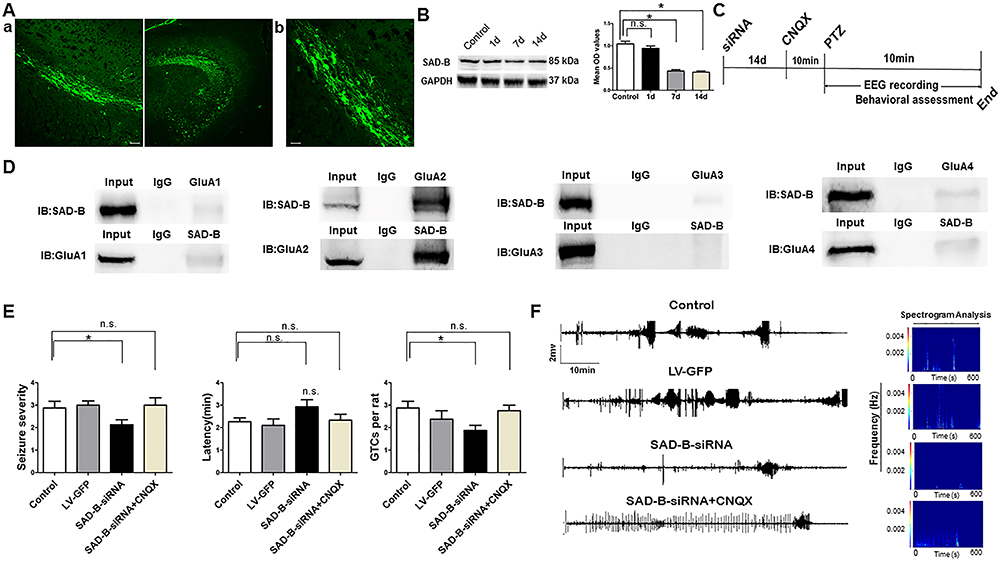
Subsequently, to explore the function of SAD-B in epileptic seizure, we downregulated SAD-B expression and observed the behavior of the PTZ-induced epileptic animals. We administered SAD-B-siRNA into the hippocampus of the animal models and observed behavior. After the injection of the exogenous recombinant lentivirus, we first detected the effect of siRNA transfection in the hippocampus 14 days later. We visualized GFP-positive cells in the hippocampus 14 days later in control animals as well (Figure 3A). Moreover, immunoblotting showed that SAD-B expression was significantly decreased in the hippocampus 7 days and 14 days after the initial injection (Figure 3B), indicating that the recombinant lentivirus was successfully transfected into the neurons in the hippocampus. Fourteen days after the administration of the lentiviral vector, we evaluated behavior following PTZ-induced seizures. Seizure severity and susceptibility were significantly reduced (lower seizure severity, fewer GTCs per rat) in the SAD-B-siRNA group compared to the control group (Figure 3E). Furthermore, multichannel hippocampal EEG recordings showed that the seizure severity and susceptibility were less intense in the SAD-B-siRNA group than in the control group (Figure 3F). These findings indicated that SAD-B was involved in epileptic seizure and that SAD-B downregulation can attenuate seizure severity and susceptibility in the PTZ-induced epileptic model.

The antiepileptic effect of brain-specific serine/threonine-protein kinase 1 (SAD-B) downregulation is mediated by AMPA receptors. A, Fluorescence (FITC)-labelled cells in the hippocampus of the rat brain showed fluorescent neurons that were transfected with exogenous LV-GFP. a: Scale bar: 50 µm (200×). b: Scale bar: 50 µm (400×). B, Immunoblot analysis showing that SAD-B expression was significantly decreased in the hippocampus 7 and 14 days after SAD-B-siRNA injection compared to controls (n=4 in each, *P<0.05, one-way ANOVA). C, Schematic overview of SAD-B-siRNA+CNQX administration in vivo. D, Representative western blotting images of co-immunoprecipitation showing a positive interaction between SAD-B and GluA2 in the hippocampi of epileptic rats. E, Behavioral evaluations showing that seizure severity and GTCs per rat were reduced in the SAD-B-siRNA group compared with the control group and that the latency was prolonged in the SAD-B-siRNA group compared to the control group (n=8 in each, *P<0.05). However, seizure severity and GTCs per rat were not reduced in the SAD-B-siRNA+CNQX group compared to the control group, and the latency was not prolonged in the SAD-B-siRNA+CNQX group compared to the control group, suggesting that the antiepileptic effect of SAD-B downregulation was abolished by CNQX. Statistical significance was evaluated by one-way ANOVA. If the variance was homogeneous, the LSD method was used to make multiple comparisons of the mean of each group; when the variance was not homogeneous, the Kruskal-Wallis test and Dunnett's T3 method were used. F, Hippocampal EEG recordings and spectrogram analysis of the electrographic results showing that seizure severity and susceptibility were less intense in the SAD-B-siRNA group than in the control group; this effect was abolished by CNQX. Data are reported as mean±SE. n.s., not significant.
The antiepileptic effect of SAD-B downregulation was mediated by AMPA receptors
SAD-B regulates AMPA receptor-mediated neurotransmission through a presynaptic mechanism (88. Inoue E, Mochida S, Takagi H, Higa S, Deguchi-Tawarada M, Takao-Rikitsu E, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron 2006; 50: 261–275, doi: 10.1016/j.neuron.2006.03.018.
https://doi.org/10.1016/j.neuron.2006.03...
). Thus, we explored the possibility that this mechanism is involved in epilepsy. We found that SAD-B interacted with GluA2 (AMPA receptors are comprised of four subunits (GluA1-GluA4) (Figure 3D). Animals were treated with siRNA or vehicle (14 days prior to receiving PTZ), and then with CNQX (0.5 μg/kg, 10 min prior to receiving PTZ). However, when AMPA receptors were blocked by CNQX, SAD-B downregulation no longer attenuated seizure severity and susceptibility in the PTZ-induced model (Figure 3E). Moreover, multichannel hippocampal EEG recordings also showed that the seizure severity and susceptibility were not attenuated in the SAD-B-siRNA+CNQX group compared to the control group (Figure 3F). These findings indicated that the antiepileptic effect of SAD-B downregulation in PTZ-induced epilepsy was likely mediated through AMPA receptors.
Discussion
Epilepsy is one of the most common neurological disorders in the nervous system and its pathophysiological alterations result in abnormal discharges characterized by the periodic and unpredictable occurrence of seizures. Epilepsy has many causes (e.g., genetic, developmental, and acquired), each of which may lead to abnormal neuronal synchronization. Cascading excitation within networks of synaptically connected excitatory glutamatergic neurons plays a major role in epilepsy (44. Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr 2011; 11: 56–63, doi: 10.5698/1535-7511-11.2.56.
https://doi.org/10.5698/1535-7511-11.2.5...
). AMPA receptors, the predominant mediators of glutamate-induced excitatory neurotransmission, mediate fast neurotransmission at the postsynaptic site. AMPA receptors are critical for the generation and spread of epileptic activity (44. Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr 2011; 11: 56–63, doi: 10.5698/1535-7511-11.2.56.
https://doi.org/10.5698/1535-7511-11.2.5...
), and evidence has illustrated that AMPA receptors show hypersensitivity in human hippocampal and neocortical tissue in the brains of patients with epilepsy (1212. Zilles K, Qü MS, Köhling R, Speckmann EJ. Ionotropic glutamate and GABA receptors in human epileptic tissue: quantitative in vitro receptor autoradiography. Neuroscience 1999; 94: 1051–1061, doi: 10.1016/S0306-4522(99)00392-9.
https://doi.org/10.1016/S0306-4522(99)00...
,1313. Vollmar W, Gloger J, Berger E. RNA editing (R/G site) and flip-flop splicing of the AMPA receptor subunit GluR2 in nervous tissue of epilepsy patients. Neurobiol Dis 2004; 15: 371–379, doi: 10.1016/j.nbd.2003.11.006.
https://doi.org/10.1016/j.nbd.2003.11.00...
). The upregulation of AMPA receptors is associated with audiogenic seizures and pharmacoresistant focal temporal lobe epilepsy (1414. Palomero-Gallagher N, Schleicher A, Bidmon HJ, Pannek HW, Hans V, Gorji A, et al. Multi-receptor analysis in human neocortex reveals complex alterations of receptor ligand binding in focal epilepsies. Epilepsia 2012; 53: 1987–1997, doi: 10.1111/j.1528-1167.2012.03634.x.
https://doi.org/10.1111/j.1528-1167.2012...
,1515. Gidal BE, Laurenza A, Hussein Z, Yang H, Fain R, Edelstein J, et al. Perampanel efficacy and tolerability with enzymeinducing AEDs in patients with epilepsy. Neurology 2015; 84: 1972–1980, doi: 10.1212/WNL.0000000000001558.
https://doi.org/10.1212/WNL.000000000000...
). In an epileptic animal model, AMPA receptor phosphorylation was shown to be involved in a pilocarpine model of epilepsy, and alterations in AMPA receptor binding and subunit mRNA expression leads to generalized PTZ-induced seizures (1616. Ekonomou A, Smith AL, Angelatou F. Changes in AMPA receptor binding and subunit messenger RNA expression in hippocampus and cortex in the pen tylenetetrazole-induced 'kindling' model of epilepsy. Brain Res Mol Brain Res 2001; 95: 27–35, doi: 10.1016/S0169-328X(01)00230-3.
https://doi.org/10.1016/S0169-328X(01)00...
,1717. Lopes MW, Soares FM, de Mello N, Nunes JC, Cajado AG, de Brito D, et al. Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp Brain Res 2013; 226: 153–163, doi: 10.1007/s00221-013-3421-8.
https://doi.org/10.1007/s00221-013-3421-...
). Notably, evidence has shown that AMPA receptor agonists initiate seizures and that AMPA receptor antagonists induce antiepileptic activity (1818. Russo E, Gitto R, Citraro R, Chimirri A, De Sarro G. New AMPA antagonists in epilepsy. Expert Opin Investig Drugs 2012; 21: 1371–1389, doi: 10.1517/13543784.2012.705277.
https://doi.org/10.1517/13543784.2012.70...
). Thus, AMPA receptor blockade was applied, and it should have a promising therapeutic anticonvulsant potential.
SAD-B belongs to the AMPK-related subfamily of protein kinases and contains a conserved kinase domain (KD), an adjacent ubiquitin-associated (UBA) domain, and a kinase-associated domain 1 (KA1) (1919. Jaleel M, Villa F, Deak M, Toth R, Prescott AR, Van Aalten DM, et al. The ubiquitin-associated domain of AMPK-related kinases regulates conformation and LKB1-mediated phosphorylation and activation. Biochem J 2006; 394: 545–555, doi: 10.1042/BJ20051844.
https://doi.org/10.1042/BJ20051844...
). In this study, we demonstrated that SAD-B is involved in epileptic seizure by regulating AMPA receptor function in the PTZ-induced epileptic model. We first identified that SAD-B is located in the cortex and hippocampus of epileptic rats, in which hippocampal sclerosis is the most important pathological characteristic of epilepsy (2020. Blumcke I, Thom M, Wiestler OD. Ammon's horn sclerosis: a maldevelopmental disorder associated with temporal lobe epilepsy. Brain Pathol 2002; 12: 199–211, doi: 10.1111/j.1750-3639.2002.tb00436.x.
https://doi.org/10.1111/j.1750-3639.2002...
,2121. Novy J, Belluzzo M, Caboclo LO, Catarino CB, Yogarajah M, Martinian L, et al. The lifelong course of chronic epilepsy: the Chalfont experience. Brain 2013; 136: 3187–3199, doi: 10.1093/brain/awt117.
https://doi.org/10.1093/brain/awt117...
). This result was consistent with a previous study that indicated that SAD-B is localized in cultured rat primary hippocampal neurons (2222. Lilley BN, Krishnaswamy A, Wang Z, Kishi M, Frank E, Sanes JR. SAD kinases control the maturation of nerve terminals in the mammalian peripheral and central nervous systems. Proc Natl Acad Sci USA 2014; 111: 1138–1143, doi: 10.1073/pnas.1321990111.
https://doi.org/10.1073/pnas.1321990111...
). Subsequently, we showed that SAD-B expression was increased in patients with TLE, which indicated that SAD-B may be involved in epilepsy. To further explore the mechanism underlying this alteration and the role of SAD-B in epilepsy, we established the PTZ-induced model, which is the most widely accepted animal model for studying the effects of novel antiepileptic molecules (1111. Nutt DJ, Cowen PJ, Batts CC, Grahame-Smith DG, Green AR. Repeated administration of subconvulsant doses of GABA antagonist drugs. Psychopharmacology 1982; 76: 84–87, doi: 10.1007/BF00430762.
https://doi.org/10.1007/BF00430762...
). We found that SAD-B expression was also increased in the PTZ-induced model, which is consistent with the results in patients with TLE. These findings suggested that SAD-B may exhibit correlations with epileptic seizure. In nervous system diseases, there is evidence that SAD-B plays multiple sequential roles in neurons, including neuronal polarization, the control of axonal arborization, differentiation, specification, and the promotion of the maturation of nerve terminals (66. Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, et al. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 2007; 129: 549–563, doi: 10.1016/j.cell.2007.03.025.
https://doi.org/10.1016/j.cell.2007.03.0...
,88. Inoue E, Mochida S, Takagi H, Higa S, Deguchi-Tawarada M, Takao-Rikitsu E, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron 2006; 50: 261–275, doi: 10.1016/j.neuron.2006.03.018.
https://doi.org/10.1016/j.neuron.2006.03...
,2222. Lilley BN, Krishnaswamy A, Wang Z, Kishi M, Frank E, Sanes JR. SAD kinases control the maturation of nerve terminals in the mammalian peripheral and central nervous systems. Proc Natl Acad Sci USA 2014; 111: 1138–1143, doi: 10.1073/pnas.1321990111.
https://doi.org/10.1073/pnas.1321990111...
–2626. Lilley BN, Pan YA, Sanes JR. SAD kinases sculpt axonal arbors of sensory neurons through long- and short-term responses to neurotrophin signals. Neuron 2013; 79: 39–53, doi: 10.1016/j.neuron.2013.05.017.
https://doi.org/10.1016/j.neuron.2013.05...
). SAD-B mutants limit the localization of synaptic vesicle proteins to axons, organize them at synapses, and regulate synaptic vesicle distribution and the development of normal synapses in C. elegans (77. Crump JG, Zhen M, Jin Y, Bargmann CI. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron 2011; 29: 115–129, doi: 10.1016/S0896-6273(01)00184-2.
https://doi.org/10.1016/S0896-6273(01)00...
,2727. Hung W, Hwang C, Po MD, Zhen M. Neuronal polarity is regulated by a direct interaction between a scaffolding protein, Neurabin, and a presynaptic SAD-1 kinase in Caenorhabditis elegans. Development 2007; 134: 237–249, doi: 10.1242/dev.02725.
https://doi.org/10.1242/dev.02725...
). Tsc1/Tsc2 promotes axonal growth via the upregulation of SAD kinase in tuberous sclerosis complex, which is characterized by tumor predisposition and neurological abnormalities, including epilepsy, mental retardation, and autism (2828. Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev 2008; 22: 2485–2495, doi: 10.1101/gad.1685008.
https://doi.org/10.1101/gad.1685008...
). SAD-B knock-out mice exhibit defects in neuronal polarity and die 2 h after birth; the embryos show visibly thinner cortices and their neurons lack distinct axonal and dendritic processes (2323. Kishi M, Pan YA, Crump JG, Sanes JR. Mammalian SAD kinases are required for neuronal polarization. Science 2005; 307: 929–932, doi: 10.1126/science.1107403.
https://doi.org/10.1126/science.1107403...
24. Hagiwara A, Harada K, Hida Y, Kitajima I, Ohtsuka T. Distribution of serine/threonine kinase SAD-B in mouse peripheral nerve synapse. Neuroreport 2011; 22: 319–325, doi: 10.1097/WNR.0b013e328346013c.
https://doi.org/10.1097/WNR.0b013e328346...
25. Watabe AM, Nagase M, Hagiwara A, Hida Y, Tsuji M, Ochiai T, et al. SAD-B kinase regulates pre-synaptic vesicular dynamics at hippocampal Schaffer collateral synapses and affects contextual fear memory. J Neurochem 2016; 136: 36–47, doi: 10.1111/jnc.13379.
https://doi.org/10.1111/jnc.13379...
). Most importantly, SAD-B has been characterized as a presynaptic kinase that associates with synaptic vesicles at the active zone cytomatrix of presynaptic terminals and regulates presynaptic AMPA release (88. Inoue E, Mochida S, Takagi H, Higa S, Deguchi-Tawarada M, Takao-Rikitsu E, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron 2006; 50: 261–275, doi: 10.1016/j.neuron.2006.03.018.
https://doi.org/10.1016/j.neuron.2006.03...
). In this study, we showed that SAD-B blockade attenuated PTZ-induced epileptiform seizures and that the anticonvulsant effect of SAD-B blockade was mediated by AMPA receptors.
Using co-immunoprecipitation, we found that SAD-B interacted with GluA2, which is one of the subunits of AMPA receptors in the brain. Its interaction with GluA2 suggests that it may cause epilepsy. In the nervous system, 90–95% of AMPA receptors are GluA1/GluA2 heteromers (2929. Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, et al. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 2009; 62: 254–268, doi: 10.1016/j.neuron.2009.02.027.
https://doi.org/10.1016/j.neuron.2009.02...
,3030. Reimers JM, Milovanovic M, Wolf ME. Quantitative analysis of AMPA receptor subunit composition in addiction-related brain regions. Brain Res 2011; 1367: 223–233, doi: 10.1016/j.brainres.2010.10.016.
https://doi.org/10.1016/j.brainres.2010....
). Moreover, approximately 80% of synaptic AMPA receptors in the CA1 region of the hippocampus are GluA1/GluA2 heteromers (29. Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, et al. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 2009; 62: 254–268, doi: 10.1016/j.neuron.2009.02.027.
https://doi.org/10.1016/j.neuron.2009.02...
30. Reimers JM, Milovanovic M, Wolf ME. Quantitative analysis of AMPA receptor subunit composition in addiction-related brain regions. Brain Res 2011; 1367: 223–233, doi: 10.1016/j.brainres.2010.10.016.
https://doi.org/10.1016/j.brainres.2010....
3131. Wenthold RJ, Petralia RS, Blahos J II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CAllCA2 neurons. Neuroscience 1996; 16: 1982–1989, doi: 10.1523/JNEUROSCI.16-06-01982.1996.
https://doi.org/10.1523/JNEUROSCI.16-06-...
). Increasing evidence has indicated that GluA2 expression is reduced in a model of status epilepticus and acquired epilepsy (3232. Sommer C, Roth SU, Kiessling M. Kainate-induced epilepsy alters protein expression of AMPA receptor subunits GluR1, GluR2 and AMPA receptor binding protein in the rat hippocampus. Acta Neuropathol 2001; 101: 460–468, doi: 10.1007/s004010000310.
https://doi.org/10.1007/s004010000310...
). Furthermore, in pilocarpine-induced SE, the GluA2 reduction may last for several weeks (3333. Porter BE, Cui XN, Brooks-Kayal AR. Status epilepticus differentially alters AMPA and kainate receptor subunit expression in mature and immature dentate granule neurons. Eur J Neurosci 2006; 23: 2857–2863, doi: 10.1111/j.1460-9568.2006.04839.x.
https://doi.org/10.1111/j.1460-9568.2006...
,3434. Hu Y, Jiang L, Chen H, Zhang X. Expression of AMPA receptor subunits in hippocampus after status convulsion. Childs Nerv Syst 2012; 28: 911–918, doi: 10.1007/s00381-012-1747-3.
https://doi.org/10.1007/s00381-012-1747-...
). GluA2-lacking AMPARs play an important role in the enhancement of responses to Glu in epileptic patients (3535. Rakhade SN, Zhou C, Aujla PK, Fishman R, Sucher NJ, Jensen FE. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J Neurosci 2008; 28: 7979–7990, doi: 10.1523/JNEUROSCI.1734-08.2008.
https://doi.org/10.1523/JNEUROSCI.1734-0...
). Therefore, the GluA2 heteromers constitute the most prominent AMPA receptors in pyramidal synapses (3636. Shinohara Y. Quantification of postsynaptic density proteins: glutamate receptor subunits and scaffolding proteins. Hippocampus 2012; 22: 942–953, doi: 10.1002/hipo.20950.
https://doi.org/10.1002/hipo.20950...
). They mediate fast excitatory synaptic neurotransmission in the nervous system and play an essential role in synaptic plasticity (3737. Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca (2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci 1997; 20: 464–470, doi: 10.1016/S0166-2236(97)01100-4.
https://doi.org/10.1016/S0166-2236(97)01...
).
Drug treatment is ineffective for many patients and surgical treatment is only performed if the benefits outweigh the risks (such as hippocampal sclerosis). For these reasons, new treatments complementary to pharmacological therapy need to be developed. AMPA receptor blockade should have a promising therapeutic anticonvulsant potential (44. Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr 2011; 11: 56–63, doi: 10.5698/1535-7511-11.2.56.
https://doi.org/10.5698/1535-7511-11.2.5...
,3838. Meldrum B, Rogawski M. Molecular targets for antiepileptic drug treatment. Neurotherapeutics 2007; 4: 18–61, doi: 10.1016/j.nurt.2006.11.010.
https://doi.org/10.1016/j.nurt.2006.11.0...
). In this study, we showed that SAD-B downregulation attenuated seizure severity and susceptibility by regulating AMPA receptors in patients with TLE and in the PTZ-induced model. The role of SAD-B in the regulation of AMPA receptor function indicated that SAD-B may be a desirable target for developing therapeutic strategies aimed at epilepsy. However, the exact molecular mechanism of the SAD-B signaling pathway in seizure progression needs to be further elucidated. Whether SAD-B is involved in seizure progression through other processes, such as controlling axonal arborization and promoting the maturation of nerve terminals, needs to be further explored.
Acknowledgments
We would like to thank the First Affiliated Hospital of Chongqing Medical University for supplying the patients. We sincerely thank the patients and their families for their participation in this study. We also acknowledge the National Institutes of Health of China and the Ethics Committee on Human Research of Chongqing Medical University. In addition, we thank the National Natural Science Foundation of China that funded our work (grant numbers: 81571259; 81771390).
References
-
1Dixit AB, Tripathi M, Chandra PS, Banerjee J. Molecular biomarkers in drug-resistant epilepsy: facts & possibilities. Int J Surg 2016; 36: 483–491, doi: 10.1016/j.ijsu.2015.08.029.
» https://doi.org/10.1016/j.ijsu.2015.08.029 -
2Blümcke I, Aronica E, Miyata H, Sarnat HB, Thom M, Roessler K, et al. International recommendation for a comprehensive neuropathologic workup of epilepsy surgery brain tissue: a consensus Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 2016; 57: 348–358, doi: 10.1111/epi.13319.
» https://doi.org/10.1111/epi.13319 -
3Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav 2014; 37: 59–70, doi: 10.1016/j.yebeh.2014.05.031.
» https://doi.org/10.1016/j.yebeh.2014.05.031 -
4Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr 2011; 11: 56–63, doi: 10.5698/1535-7511-11.2.56.
» https://doi.org/10.5698/1535-7511-11.2.56 -
5Di Bonaventura C, Labate A, Maschio M, Meletti S, Russo E. AMPA receptors and perampanel behind selected epilepsies: current evidence and future perspectives. Expert Opin Pharmacother 2017; 18: 1751–1764, doi: 10.1080/14656566.2017.1392509.
» https://doi.org/10.1080/14656566.2017.1392509 -
6Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, et al. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 2007; 129: 549–563, doi: 10.1016/j.cell.2007.03.025.
» https://doi.org/10.1016/j.cell.2007.03.025 -
7Crump JG, Zhen M, Jin Y, Bargmann CI. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron 2011; 29: 115–129, doi: 10.1016/S0896-6273(01)00184-2.
» https://doi.org/10.1016/S0896-6273(01)00184-2 -
8Inoue E, Mochida S, Takagi H, Higa S, Deguchi-Tawarada M, Takao-Rikitsu E, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron 2006; 50: 261–275, doi: 10.1016/j.neuron.2006.03.018.
» https://doi.org/10.1016/j.neuron.2006.03.018 -
9Kim M, Campeau S, Falls WA, Davis M. Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav Neural Biol 1993; 59: 5–8, doi: 10.1016/0163-1047(93)91075-X.
» https://doi.org/10.1016/0163-1047(93)91075-X -
10Yu X, Xu T, Ou S, Yuan J, Deng J, Li R, et al. Endophilin A1 mediates seizure activity via regulation of AMPARs in a PTZ-kindled epileptic mouse model. Exp Neurol 2018; 304: 41–57, doi: 10.1016/j.expneurol.2018.02.014.
» https://doi.org/10.1016/j.expneurol.2018.02.014 -
11Nutt DJ, Cowen PJ, Batts CC, Grahame-Smith DG, Green AR. Repeated administration of subconvulsant doses of GABA antagonist drugs. Psychopharmacology 1982; 76: 84–87, doi: 10.1007/BF00430762.
» https://doi.org/10.1007/BF00430762 -
12Zilles K, Qü MS, Köhling R, Speckmann EJ. Ionotropic glutamate and GABA receptors in human epileptic tissue: quantitative in vitro receptor autoradiography. Neuroscience 1999; 94: 1051–1061, doi: 10.1016/S0306-4522(99)00392-9.
» https://doi.org/10.1016/S0306-4522(99)00392-9 -
13Vollmar W, Gloger J, Berger E. RNA editing (R/G site) and flip-flop splicing of the AMPA receptor subunit GluR2 in nervous tissue of epilepsy patients. Neurobiol Dis 2004; 15: 371–379, doi: 10.1016/j.nbd.2003.11.006.
» https://doi.org/10.1016/j.nbd.2003.11.006 -
14Palomero-Gallagher N, Schleicher A, Bidmon HJ, Pannek HW, Hans V, Gorji A, et al. Multi-receptor analysis in human neocortex reveals complex alterations of receptor ligand binding in focal epilepsies. Epilepsia 2012; 53: 1987–1997, doi: 10.1111/j.1528-1167.2012.03634.x.
» https://doi.org/10.1111/j.1528-1167.2012.03634.x -
15Gidal BE, Laurenza A, Hussein Z, Yang H, Fain R, Edelstein J, et al. Perampanel efficacy and tolerability with enzymeinducing AEDs in patients with epilepsy. Neurology 2015; 84: 1972–1980, doi: 10.1212/WNL.0000000000001558.
» https://doi.org/10.1212/WNL.0000000000001558 -
16Ekonomou A, Smith AL, Angelatou F. Changes in AMPA receptor binding and subunit messenger RNA expression in hippocampus and cortex in the pen tylenetetrazole-induced 'kindling' model of epilepsy. Brain Res Mol Brain Res 2001; 95: 27–35, doi: 10.1016/S0169-328X(01)00230-3.
» https://doi.org/10.1016/S0169-328X(01)00230-3 -
17Lopes MW, Soares FM, de Mello N, Nunes JC, Cajado AG, de Brito D, et al. Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp Brain Res 2013; 226: 153–163, doi: 10.1007/s00221-013-3421-8.
» https://doi.org/10.1007/s00221-013-3421-8 -
18Russo E, Gitto R, Citraro R, Chimirri A, De Sarro G. New AMPA antagonists in epilepsy. Expert Opin Investig Drugs 2012; 21: 1371–1389, doi: 10.1517/13543784.2012.705277.
» https://doi.org/10.1517/13543784.2012.705277 -
19Jaleel M, Villa F, Deak M, Toth R, Prescott AR, Van Aalten DM, et al. The ubiquitin-associated domain of AMPK-related kinases regulates conformation and LKB1-mediated phosphorylation and activation. Biochem J 2006; 394: 545–555, doi: 10.1042/BJ20051844.
» https://doi.org/10.1042/BJ20051844 -
20Blumcke I, Thom M, Wiestler OD. Ammon's horn sclerosis: a maldevelopmental disorder associated with temporal lobe epilepsy. Brain Pathol 2002; 12: 199–211, doi: 10.1111/j.1750-3639.2002.tb00436.x.
» https://doi.org/10.1111/j.1750-3639.2002.tb00436.x -
21Novy J, Belluzzo M, Caboclo LO, Catarino CB, Yogarajah M, Martinian L, et al. The lifelong course of chronic epilepsy: the Chalfont experience. Brain 2013; 136: 3187–3199, doi: 10.1093/brain/awt117.
» https://doi.org/10.1093/brain/awt117 -
22Lilley BN, Krishnaswamy A, Wang Z, Kishi M, Frank E, Sanes JR. SAD kinases control the maturation of nerve terminals in the mammalian peripheral and central nervous systems. Proc Natl Acad Sci USA 2014; 111: 1138–1143, doi: 10.1073/pnas.1321990111.
» https://doi.org/10.1073/pnas.1321990111 -
23Kishi M, Pan YA, Crump JG, Sanes JR. Mammalian SAD kinases are required for neuronal polarization. Science 2005; 307: 929–932, doi: 10.1126/science.1107403.
» https://doi.org/10.1126/science.1107403 -
24Hagiwara A, Harada K, Hida Y, Kitajima I, Ohtsuka T. Distribution of serine/threonine kinase SAD-B in mouse peripheral nerve synapse. Neuroreport 2011; 22: 319–325, doi: 10.1097/WNR.0b013e328346013c.
» https://doi.org/10.1097/WNR.0b013e328346013c -
25Watabe AM, Nagase M, Hagiwara A, Hida Y, Tsuji M, Ochiai T, et al. SAD-B kinase regulates pre-synaptic vesicular dynamics at hippocampal Schaffer collateral synapses and affects contextual fear memory. J Neurochem 2016; 136: 36–47, doi: 10.1111/jnc.13379.
» https://doi.org/10.1111/jnc.13379 -
26Lilley BN, Pan YA, Sanes JR. SAD kinases sculpt axonal arbors of sensory neurons through long- and short-term responses to neurotrophin signals. Neuron 2013; 79: 39–53, doi: 10.1016/j.neuron.2013.05.017.
» https://doi.org/10.1016/j.neuron.2013.05.017 -
27Hung W, Hwang C, Po MD, Zhen M. Neuronal polarity is regulated by a direct interaction between a scaffolding protein, Neurabin, and a presynaptic SAD-1 kinase in Caenorhabditis elegans. Development 2007; 134: 237–249, doi: 10.1242/dev.02725.
» https://doi.org/10.1242/dev.02725 -
28Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev 2008; 22: 2485–2495, doi: 10.1101/gad.1685008.
» https://doi.org/10.1101/gad.1685008 -
29Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, et al. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 2009; 62: 254–268, doi: 10.1016/j.neuron.2009.02.027.
» https://doi.org/10.1016/j.neuron.2009.02.027 -
30Reimers JM, Milovanovic M, Wolf ME. Quantitative analysis of AMPA receptor subunit composition in addiction-related brain regions. Brain Res 2011; 1367: 223–233, doi: 10.1016/j.brainres.2010.10.016.
» https://doi.org/10.1016/j.brainres.2010.10.016 -
31Wenthold RJ, Petralia RS, Blahos J II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CAllCA2 neurons. Neuroscience 1996; 16: 1982–1989, doi: 10.1523/JNEUROSCI.16-06-01982.1996.
» https://doi.org/10.1523/JNEUROSCI.16-06-01982.1996 -
32Sommer C, Roth SU, Kiessling M. Kainate-induced epilepsy alters protein expression of AMPA receptor subunits GluR1, GluR2 and AMPA receptor binding protein in the rat hippocampus. Acta Neuropathol 2001; 101: 460–468, doi: 10.1007/s004010000310.
» https://doi.org/10.1007/s004010000310 -
33Porter BE, Cui XN, Brooks-Kayal AR. Status epilepticus differentially alters AMPA and kainate receptor subunit expression in mature and immature dentate granule neurons. Eur J Neurosci 2006; 23: 2857–2863, doi: 10.1111/j.1460-9568.2006.04839.x.
» https://doi.org/10.1111/j.1460-9568.2006.04839.x -
34Hu Y, Jiang L, Chen H, Zhang X. Expression of AMPA receptor subunits in hippocampus after status convulsion. Childs Nerv Syst 2012; 28: 911–918, doi: 10.1007/s00381-012-1747-3.
» https://doi.org/10.1007/s00381-012-1747-3 -
35Rakhade SN, Zhou C, Aujla PK, Fishman R, Sucher NJ, Jensen FE. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J Neurosci 2008; 28: 7979–7990, doi: 10.1523/JNEUROSCI.1734-08.2008.
» https://doi.org/10.1523/JNEUROSCI.1734-08.2008 -
36Shinohara Y. Quantification of postsynaptic density proteins: glutamate receptor subunits and scaffolding proteins. Hippocampus 2012; 22: 942–953, doi: 10.1002/hipo.20950.
» https://doi.org/10.1002/hipo.20950 -
37Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca (2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci 1997; 20: 464–470, doi: 10.1016/S0166-2236(97)01100-4.
» https://doi.org/10.1016/S0166-2236(97)01100-4 -
38Meldrum B, Rogawski M. Molecular targets for antiepileptic drug treatment. Neurotherapeutics 2007; 4: 18–61, doi: 10.1016/j.nurt.2006.11.010.
» https://doi.org/10.1016/j.nurt.2006.11.010
-
Erratum
Rong Lihttps://orcid.org/0000-0002-4465-33241, Miaoqing Hehttps://orcid.org/0000-0001-7116-09822,3, Bing Wuhttps://orcid.org/0000-0002-7153-41364, Peng Zhanghttps://orcid.org/0000-0001-8071-37291, Qinbin Zhanghttps://orcid.org/0000-0002-9772-15111, and Yangmei Chenhttps://orcid.org/0000-0002-0560-829911Department of Neurology, Second Affiliated Hospital of Chongqing Medical University, Chongqing, China2Center for Brain Disorders Research, Capital Medical University, Feng Tai District, Beijing, China3Beijing Institute for Brain Disorders, Feng Tai District, Beijing, China4Department of Neurology, First Affiliated Hospital of Chongqing Medical University, Chongqing Key Laboratory of Neurology, Chongqing, ChinaCorrespondence: Yangmei Chen: <chenym1997@cqmu.edu.cn>Erratum for: Braz J Med Biol Res | doi: 10.1590/1414-431X20199175 | PMID: 32267308 | PMCID: PMC7162585On July 7, 2020, the Brazilian Journal of Medical and Biological Research received a request from the first author Rong Li requesting the substitution of Figure 1B, Panels: Rat Hippocampus SAD-B, MAP2, and MERGE because these 3 images had been submitted incorrectly. This modification of Figure 1 does not change the findings of this research. After careful evaluation by the Editors, this erratum is being published. Figure 1
Figure 1
Brain-specific serine/threonine-protein kinase 1 (SAD-B) is localized in the epileptic brain. A, Immunofluorescence labelling of SAD-B (red), MAP2 (violet), and GFAP (green) in the cortex of patients with temporal lobe epilepsy (TLE) showing that SAD-B was co-localized with MAP2 but not with GFAP. Scale bar: 50 μm (400×). B, Immunofluorescence labelling of SAD-B (red), MAP2 (violet), and GFAP (green) in the CA1 region of the hippocampus or cortex of an epileptic rat showing that SAD-B was co-localized with MAP2, but not with GFAP. Scale bar: 50 μm (400×). White arrows: SAD-B; yellow arrows: GFAP.
Publication Dates
-
Publication in this collection
06 Apr 2020 -
Date of issue
2020
History
-
Received
31 July 2019 -
Accepted
20 Dec 2019