Abstracts
In this paper we report the application of liquid chromatography-mass spectrometry (LC-MS-MS) to the identification of the products formed by oxidation of albendazole and disopyramide with metalloporphyrins in dichloroethane, using iodosylbenzene as an oxygen donor. Our results show that LC-MS-MS is a powerful tool to study the in vitro metabolism of drugs, allowing the identification of known and unknown metabolites. In addition, it was observed that the catalyst system used resulted in the formation of the same metabolites as obtained in vivo, although for disopyramide other products were also observed.
LC-MS-MS; metabolism; metalloporphyrin; disopyramide; albendazole
Neste artigo estamos relatando a aplicação da cromatografia líquida de alta eficiência acoplada à espectrometria de massas (LC-MS-MS) para a identificação dos produtos formados na oxidação de albendazol e disopiramida utilizando metaloporfirinas em dicloroetano como catalisador e iodozilbenzeno como doador de oxigênio. Nossos resultados comprovam que LC-MS-MS é uma poderosa técnica para o estudo in vitro de metabolismo de fármacos, permitindo a identificação de metabólitos conhecidos ou não. Foi observado que o sistema catalítico usado resultou na formação dos mesmos metabólitos obtidos in vivo, embora outros produtos também foram formados na oxidação da disopiramida.
ARTICLE
LC-MS-MS identification of drug metabolites obtained by metalloporphyrin mediated oxidation
Andrea J. M. MaurinI; Yassuko IamamotoII; Norberto P. LopesI; John R. Lindsay-SmithIII; Pierina S. BonatoI
IDepartamento de Física e Química, Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Av. do Café, s/n, 14040-903 Ribeirão Preto - SP, Brazil
IIDepartamento de Química, Faculdade de Filosofia Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, Av. Bandeirantes, 3900, 14040-901 Ribeirão Preto - SP, Brazil
IIIDepartment of Chemistry, University of York, York, United Kingdom
Correspondence Correspondence to Pierina S. Bonato e-mail: psbonato@fcfrp.usp.br Received: October 19, 2002 Published on the web: March 28, 2003 FAPESP helped in meeting the publication costs of this article
ABSTRACT
In this paper we report the application of liquid chromatography-mass spectrometry (LC-MS-MS) to the identification of the products formed by oxidation of albendazole and disopyramide with metalloporphyrins in dichloroethane, using iodosylbenzene as an oxygen donor. Our results show that LC-MS-MS is a powerful tool to study the in vitro metabolism of drugs, allowing the identification of known and unknown metabolites. In addition, it was observed that the catalyst system used resulted in the formation of the same metabolites as obtained in vivo, although for disopyramide other products were also observed.
Keywords: LC-MS-MS, metabolism, metalloporphyrin, disopyramide, albendazole
RESUMO
Neste artigo estamos relatando a aplicação da cromatografia líquida de alta eficiência acoplada à espectrometria de massas (LC-MS-MS) para a identificação dos produtos formados na oxidação de albendazol e disopiramida utilizando metaloporfirinas em dicloroetano como catalisador e iodozilbenzeno como doador de oxigênio. Nossos resultados comprovam que LC-MS-MS é uma poderosa técnica para o estudo in vitro de metabolismo de fármacos, permitindo a identificação de metabólitos conhecidos ou não. Foi observado que o sistema catalítico usado resultou na formação dos mesmos metabólitos obtidos in vivo, embora outros produtos também foram formados na oxidação da disopiramida.
Introduction
During the development of a new drug, several preclinical studies may be conducted in order to establish the real potentiality of this drug candidate, i.e, if the drug has acceptable absorption, distribution, metabolism, and excretion properties. These studies include assays to predict or measure the solubility of the compound, metabolic pathway, protein binding, enzyme induction, drug-drug interaction, etc.1 Among them, drug metabolism is of major importance because metabolism can significantly affect the drug's safety and efficacy due to the formation of therapeutically active or toxic metabolites.
In addition, drugs already marketed have also been subjected to metabolism studies in order to obtain additional information. An example of this situation is the study of the effect of stereoselectivity on drug metabolism, an issue that was not considered when several drugs were developed and marketed as racemates in the past.2,3
Several in vitro models have been used to study drug metabolism such as subcellular fractions, hepatocytes, liver slices and, more recently, recombinant enzymes.4 Another direction in the study of drug metabolism is the use of a chemical catalyst to mimic the oxidative biotransformation of drugs usually performed by cytochrome P-450 or peroxidases, due to the easier preparation, purification and characterization of potentially reactive metabolites because of the absence of proteins in the reaction mixture.5 Since cytochrome P-450 and many peroxidases contain an iron (III) protoporphyrin IX as a prosthetic group, many attempts have been made to mimic their oxygenase and oxidase activities with synthetic metalloporphyrins.6-8
Regardless of the model used to study drug metabolism, identification of the metabolites plays an important role in the process. Therefore, the development of new technologies capable of generating rapid and accurate elucidation of these metabolites has become essential for this kind of study.
High-performance liquid chromatography (HPLC) coupled on-line to mass spectrometry (MS) combines the advantages of both techniques, i.e., the high selectivity and separation efficiency of chromatography and the structural information and further increase in selectivity of mass spectrometry.9,10 In addition, coupling LC to MS is relatively straightforward to accomplish, due to the development of atmospheric pressure ionization sources, such as electrospray and atmospheric pressure chemical ionization. These ionization sources are relatively soft and produce mainly the molecular ion. Further structural information as required for metabolic studies can be obtained by combining high performance LC with tandem MS, in triple stage quadrupole or ion-trap mass spectrometers (LC-MS-MS or LC-MSn).9,11
In a triple stage quadrupole mass spectrometer, the identification of the metabolites can be carried out in the linear scan mode (MS) or daughter scan mode (MS-MS). Other possibilities include parent ion scanning, used when there is the need to determine or to ascertain the origin of a particular product ion, selected reaction monitoring, for those cases where the analyte of interest is known or expected, and constant neutral loss scanning, a mode suited to samples where more than one metabolite sharing the same metabolic pathway is expected.11 In the ion-trap instrument, multiple stages of MS-MS can be applied to achieve structure elucidation of the unknown.9
This paper describes the use of this technique to identify the products obtained by oxidation of two drugs using metalloporphyrins (Figure 1) as catalysts. The drugs selected for this study were disopyramide (DP) and albendazole (ABZ). Although their metabolic pathways are known, this is the first report showing the use of metalloporphirins as a catalyst system. The oxidation products formed were identified based on their MS fragmentation pattern and also by comparing the retention times for those metabolites for which we have authentic standards in our laboratory.
Material and Methods
Chemicals and reagents
DP and mono-N-depropyldisopyramide (MNDP) were kindly supplied by Laboratórios Silva Araújo Roussel Roussel UCLAF (Brazil). ABZ, albendazole sulfoxide (ASOX) and albendazole sulfone (ASON) were provided by Robert Young & Co. Ltd (Scotland, U.K.). Iodosylbenzene (PhIO) was synthesized according to a previously described method.12 The porphyrins H2(TDCPP) and H2(TNPP) (Figure 1) were purchased from Midcentury (USA) and iron insertion into the free base porphyrins was carried out according to the literature.13 The perchloration of [Fe(TNPP)] was carried out according to Wijesekera et al.14
Methanol and acetonitrile (EM Science, USA) were of HPLC grade. All other chemicals were analytical-reagent grade and were used without further purification. The water used for mobile phase preparation was purified with a Milli-Q Plus System (Millipore, USA).
Oxidation reactions
The reaction mixtures (2 mL) in dichloroethane contained 6.0 mmol L-1 DP or ABZ, 0.3 mmol L-1 metalloporphyrin and 9.0 mmol L-1 PhIO. All experiments were carried out at room temperature, under an air atmosphere, in a glass vessel equipped with a magnetic stirring bar. Reaction times were measured after the addition of PhIO. At specified intervals, the magnetic stirring was stopped and an aliquot of the reaction mixture (10 mL) was withdrawn. After the addition of 90 mL hexane and 100 mL mobile phase, the mixture was vortex mixed and centrifuged and the lower phase (mobile phase) was injected into the chromatographic system. This extraction procedure was carried out to eliminate the porphyrin (soluble in hexane) from the sample
Equipment and chromatographic conditions
The oxidation products of ABZ were analyzed with an HPLC system consisting of two LC10AD solvent pumps, an SLC 10A system controller, a CTO-10AS column oven (Shimadzu, Japan) and a 7125 Rheodyne injector with a 20 mL loop. A UV detector (SPD 10A, Shimadzu, Japan) set at 260 nm (ABZ) was also used. Separations were carried out at 22 oC on a LiChrospher CN column (125 x 4.6 mm I.D., 5 mm particle size, Merck, Germany). A CN guard column (4 x 4 mm I.D., Merck) was used to protect the analytical column. The mobile phase for the analysis of ABZ and its metabolites consisted of methanol:water (4:6, v/v) acidified with 1% acetic acid, at a flow rate of 1 mL min-1.
The MS system was a Quatro LC triple-stage quadrupole (Micromass, United Kingdom), fitted with a Z-electrospray interface operating in the positive ion mode and calibrated with sodium iodide/cesium iodide in the 50 to 2000 Da range. The source block and desolvation temperatures were 100 oC and 250 oC, respectively. Nitrogen was used as both drying and nebulizing gas at 44 and 555 L h-1, respectively. Argon was used as collision gas. Cone and collision cell voltages were set at 30 V and 20 eV, respectively. The HPLC eluent was split by a Valco valve and a flow rate of approximately 0.1 mL min-1 was introduced into the stainless steel capillary probe. The spectra were obtained in the MS scan mode and daughter scan mode.
The LC-MS-MS system used for the analysis of DP consisted of a Varian 5000 pump, a Rheodyne 7125 injector with a 20 mL loop and a Finnigan MAT LCQ mass spectrometer (USA) equipped with an ion-trap mass spectrometer and an atmospheric pressure chemical ionization interface. The vaporizer temperature was set to 400 oC and nitrogen was applied as the sheath gas at a flow rate of 50 (arbitrary units). The heated capillary was maintained at 150 oC. Mass analysis was performed in the positive ion mode with the source current set at 5 mA, and the potentials of tube lens and capillary set at 45 and 9 V, respectively. The analyses were carried out with a laboratory-made octylsilane reversed phase column (5 mm particle size, 150 x 4 mm) endcapped with trimethylchlorosilane, protected by an RP-8 guard column (4 x 4 mm, Merck, Germany). Elution was carried out at a flow rate of 1 mL min-1 using ammonium acetate buffer, 0.1 mol L-1, pH 4.2:acetonitrile (75:25, v/v) as the mobile phase.
Results and Discussion
Characterization and identification of ABZ oxidation products
ABZ, [methyl-5-(propylthio)-1-H-benzimidazol-2-yl]carbamate, has been used for the treatment of neurocysticercosis15,16 and undergoes extensive metabolism by liver microsomal enzymes, and probably also in the gastrointestinal tract, to its major active metabolite, albendazole sulfoxide (ASOX). This metabolite is further metabolized to albendazole sulfone (ASON), which does not appear to have any activity.17,18
Figure 2 shows the total ion chromatogram referring to the analysis of the products obtained by oxidation of ABZ with [Fe(TNPCl8P)]Cl porphirin after a 1 hour reaction. The products were identified by comparing their retention times with the retention times obtained by direct injection of standard solutions of ASOX (tR = 6.8 min) and ASON (tR = 5.7 min) and also using the MS scan and daughter scan spectra (Figure 3). The peak eluting at 16.4 min refers to ABZ and its mass spectrum is also presented in Figure 3. The positive electrospray full-scan spectra of all compounds indicated that the protonated molecules ([M-H]+) were the most abundant ions, and therefore these ions were selected for the fragmentation in the collision cell, to obtain the daughter scan spectra.
Figure 4 shows the fragmentation route for ASOX. The bond between the 2p orbital of O and the 3d orbital of the S atom (in the sulfoxides) is similar to that observed in the phosphorous ylids and can be represented by two valence bond (S=O or S+-O-). Phenyl sulfoxides produce enones by heating through an elimination reaction. The elimination mechanism involves a reverse cycloaddition (pericyclic reaction) and gave phenyl sulfenic acid19. The MS-MS analysis of the protonated parent ion of ASOX (fragment A) showed fragment B as the major fragment (Figure 4, pathway 1). The low reactivity of the sulfide (ABZ) and sulfone (ASON) for the reverse cycloaddition elimination was observed in the MS-MS spectra of m/z 266 (ABZ) and m/z 298 (ASON). In both experiments the loss of 42 mass units from the parent ion was less than 1 and 5%, respectively. The remote charged elimination of methanol was initiated by the proton, inducing an increase in the electron density in the carboxyl group followed by gas-phase elimination (Figure 4). The loss of 32 mass units from the parent ion gave the major fragments in ABZ and ASON analyses. The loss of C=O was probably due to the resonance system as shown in Figure 4. Finally, during the MS experiments it was possible to observe losses of 42 or 32 mass units from ions that first lost methanol and CH2=CH2, respectively.
Based on these results we may conclude that the metalloporhyrin utilized is a biomimetic catalyst of ABZ due to the formation of the same major metabolites obtained in vivo. The time dependence profile of ABZ oxidation by [Fe(TNPCl8P)]Cl porphyrin is shown in Figure 5. The oxidation of ABZ was very fast, i.e., a yield of 97% for ASOX after a 2 min reaction. This product was then further oxidized to ASON.
Characterization and identification of DP oxidation products
DP, [4-di-isopropylamino-2-phenyl-2-(2-pyridyl) butyramide], is an effective antiarrhythmic drug used for the treatment and prophylaxis of ventricular and supraventricular arrhythmias.20 After administration to humans, DP is metabolized by the cytochrome P-450 family, producing mainly an MNDP metabolite.21,22
The HPLC chromatogram from the oxidation reaction of DP with [Fe(TDCPP)]Cl after 24 hours is shown in Figure 6. In addition to MNDP, the in vivo and in vitro metabolite of DP, other oxidation products were also detected. Unknown oxidation products were identified by LC-MSn (Table 1). The peaks corresponding to DP and MNDP were also confirmed by comparing the fragmentation pattern and the retention times of authentic standards.
Aliphatic amines normally have a pKa of 10 and are known as neutral nitrogen bases23. If the N lone pair is in a sp or sp2 hybridized orbital, the electron density on N and the pKa decrease. Pyridine (sp2, pKa = 5.2) is less basic than the amines. Amides are weak bases protonated on the O by the formation of a delocalized cation.19 The MS analysis of DP, MNDP and Product B showed protonated parent ions at m/z 340, 298 and 326, respectively. Every MS-MS experiments revealed odd molar mass fragments, indicating loss of a N. The protonations probably occured on aliphatic amines and DP fragmentation showed an amine elimination assisted by the aromatic N (fragment DP1, m/z 239, Figure 7A). The MNDP (R = H) and Product B (R = CH2CH3) MS-MS spectra exhibited the same fragments M1 and M2 (Figure 7B). The protonated secondary amines are more available (steric effect) to react with the amide than the tertiary amine (DP). In MNDP (R = H) and Product B (R = CH2CH3) the elimination occurred assisted by the O, as shown in Figure 7b. The MS3 of the fragment M1 (m/z 281) yielded the fragment M2 (m/z 239) and confirmed the proposed mechanism (Table 1).
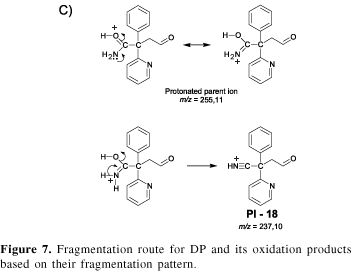
Product A has an odd parent ion indicating the replacement of a N by an O or an alkyl group. The MS analysis showed the protonated parent ion (m/z 255) and water elimination (m/z 237). As discussed before, amides are weak bases and the protonation occurs on the O. In this case, the N assistance helps to eliminate water, as shown in Figure 7C. Finally, the MS-MS analysis of the fragment PI-18 (m/z 237) yielded a fragment at m/z 237. The 14 mass unit difference in the fragment ion masses indicates the neutral elimination of C=O and is in agreement with the proposed structure.
Figure 8 shows the time dependence profile of DP oxidation using [Fe(TDCPP)]Cl porphyrin. MNDP, the in vivo metabolite, is also the main product formed in the oxidation reaction.
Conclusions
Oxidation of ABZ by [Fe(TNPCl8P)]Cl porphyrin resulted in the formation of ASOX and ASON, the major metabolites of ABZ obtained in vivo. In addition to the formation of MNDP, the in vivo metabolite of DP, two other products were also observed by oxidation of disopyramide with [Fe(TDCPP)]Cl. These unknown products were identified based on the fragmentation pattern obtained by LC-MSn . Our results show that LC-MS-MS is a powerful tool to study the in vitro metabolism of drugs, allowing the identification of known and unknown metabolites.
Acknowledgements
The authors are grateful to FAPESP (Fundação de Amparo a Pesquisa do Estado de São Paulo), CNPq (Conselho Nacional de Desenvolvimento Ciêntifico e Tecnológico), and the British Council for financial support and for research fellowships. The authors are also grateful to Profa. Dra. Isabel C. S. F. Jardim (Instituto de Química, UNICAMP), for packing the column used in the analysis of disopyramide.
- 1. Wienkers, L.C.; J. Pharmacol. Toxicol. Meth. 2001, 45, 79.
- 2. Marzo, A.; Balant, L.P.; J. Chromatogr. B 1996, 678, 73.
- 3. Caldwell, J.; J. Chromatogr. A 1995, 694, 39.
- 4. Ekins, S.; Ring, B.J.; Grace, J.; Mcrobie-Belle, D.J.; Wrighton, S.A.; J. Pharmacol. Toxicol. Meth. 2000, 44, 313.
- 5. Mansuy, D.; Pure Appl. Chem. 1994, 66, 737.
- 6. Chorghade, M.S.; Hill, D.R.; Lee, E.C.; Pariza, R.J.; Dolphin, D.H.; Hino, F.; Zhang, L.Y.; Pure Appl. Chem. 1996, 68, 753.
- 7. Vidal, M.; Bonnafous, M.; Defrance, S.; Loiseau, P.; Bernadou, J.; Meunier, B.; Drug Metabol. Dispos. 1993, 21, 811.
- 8. Bernadou, J.; Bonnafous, M.; Labat, G.; Loiseau, P.; Meunier, B.; Drug Metabol. Dispos. 1991, 19, 360.
- 9. Niessen, W.M.A.; J. Chromatogr. A 1999, 856, 179.
- 10. Vékey, K.; J. Chromatogr. A 2001, 921, 227.
- 11. Oliveira, E.J.; Watson, D.G.; Biomed. Chromatogr. 2000, 14, 351.
- 12. Sharefkin, J. G.; Saltzmann, H.; Org. Synth 1963, 43, 62.
- 13. Adler, A. D.; Longo, F. R.; Sshergalis, W.; J. Am. Chem. Soc. 1964, 86, 3145.
- 14. Wijesekera, T.; Dupre, D.; Cader, M.S.R.; Dolphin, D.; Bull Soc. Chim. Fr. 1996, 133, 765.
- 15. Del Brutto, O.H.; Arch. Neurol. 1995, 52, 102.
- 16. Del Brutto, O.H.; Sotelo, J.; Roman, G.C.; Clin. Infec. Dis. 1993, 17, 730.
- 17. Sotelo, J.; Jung, H.; Clin. Pharmacokinet 1998, 34, 503.
- 18. Villaverde, C.; Alvarez, A.I.; Redondo, P.; Voces, J.; Del Estal, J.L.; Prieto, J.G.; Xenobiotica 1995, 25, 433.
- 19. Clayden, J.; Greeves, N.; Warren, S.; Wothers, P.; Organic Chemistry, Oxford University Press: Oxford, 2001.
- 20. Hardman, J.G.; Limbird, L.E. eds.; Goodman & Gilman's the Pharmacological Basics of Therapeutics, 9th ed., McGraw-Hill: New York, 1996.
- 21. Echizen, H.; Tanizaki, M.; Tatsuno, J.; Chiba, K.; Berwick, T.; Tani, M.; Gonzalez, F.J.; Ishizaki, T.; Drug Metabol. Dispos. 2000, 28, 937.
- 22. Zhang, L.; Fitzloff, J.F.; Engel, L.C.; Cook, C.S.; Xenobiotica 2001, 31, 73.
- 23. Sykes, P.; Guia de Mecanismos da Química Orgânica, Editora da Universidade Nova de Lisboa: Lisboa, 1981.
Publication Dates
-
Publication in this collection
13 June 2003 -
Date of issue
Apr 2003
History
-
Accepted
28 Mar 2003 -
Received
19 Oct 2002