Abstracts
The electrochemical behavior of tin polycrystals in borate buffer solutions at pH 7.5 was systematically investigated using cyclic voltammetry, EC-STM and electrochemical impedance measurements. A systematic shift to more negative values of the potentials corresponding either to the major reduction peak either to the hydrogen evolution was measured when the anodization potential used for film growth was increased. A discussion about the different oxides there formed was included and a reassignation of tin oxidation and tin oxides reduction processes was given.
tin oxide; anodic films; borate buffer; gas sensors
Neste trabalho, foi estudado o comportamento eletroquímico de estanho policristalino em soluções de tampão borato com pH 7.5, utilizando voltametria cíclica, eletroquímica com microscopia de varredura (EC-STM) e espectroscopia de impedância eletroquímica. Os potenciais do pico principal de redução do óxido e de geração de hidrogênio deslocaram-se de forma sistemática para valores mais negativos quando se aumentou o potencial anódico de formação do filme. Foi discutida a natureza dos diferentes óxidos formados e também foi dada nova atribuição aos processos de oxidação do estanho e de redução de seus óxidos.
ARTICLE
An electrochemical study of tin oxide thin film in borate buffer solutions
Raül DíazI; Ismael Díez-PérezI; Pau GorostizaI; Fausto Sanz* * e-mail: sanz@qf.ub.es , I; Joan R. MoranteII
ILCTEM, Department of Physical Chemistry, University of Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain
IIEnginyeria i Materials Electrònics, Department of Electronics, University of Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain
ABSTRACT
The electrochemical behavior of tin polycrystals in borate buffer solutions at pH 7.5 was systematically investigated using cyclic voltammetry, EC-STM and electrochemical impedance measurements. A systematic shift to more negative values of the potentials corresponding either to the major reduction peak either to the hydrogen evolution was measured when the anodization potential used for film growth was increased. A discussion about the different oxides there formed was included and a reassignation of tin oxidation and tin oxides reduction processes was given.
Keywords: tin oxide, anodic films, borate buffer, gas sensors
RESUMO
Neste trabalho, foi estudado o comportamento eletroquímico de estanho policristalino em soluções de tampão borato com pH 7.5, utilizando voltametria cíclica, eletroquímica com microscopia de varredura (EC-STM) e espectroscopia de impedância eletroquímica. Os potenciais do pico principal de redução do óxido e de geração de hidrogênio deslocaram-se de forma sistemática para valores mais negativos quando se aumentou o potencial anódico de formação do filme. Foi discutida a natureza dos diferentes óxidos formados e também foi dada nova atribuição aos processos de oxidação do estanho e de redução de seus óxidos.
Introduction
Tin is a fairly corrosion-resistant metal, widely used in many applications in industry. Investigations of its electrochemical behavior (passivation, corrosion) are therefore of considerable interest, since the corrosion resistance of tin in humid atmospheres and in aqueous solutions is attributed to the presence of a passive oxide/hydroxide film on the metal surface.1
Since electrochemical processes at metallic electrodes are often affected by the presence of a surface oxide film, knowledge of the semiconducting properties of such films is necessary in order to understand their influence in film growth, charge transfer, etc. These films are most frequently non-stoichiometric oxides and of amorphous or polycrystalline structure.2 The principles of the band theory of solids have been quite successfully extended to this type of materials3 and it is known that the presence of a semiconducting oxide film determines the charge distribution and the potential drop at the metal-metal oxide-electrolyte interface, i.e., the character of the double layer. Moreover, in the case of tin the understanding of the local structural and electronic properties of these thin solid films is important in the areas of microelectronics, optical film semiconductor technology and corrosion, since SnO 2 is one of the most studied metal oxides due to its wide applications ranging from electrochemistry to optics, solar cells or gas sensor.
There are several contributions concerning the study of tin in different electrolytic media.4-11 In alkaline solutions, the anodic passivation of tin is a complex, multi-step process that is due to the presence of an oxide layer on the metal surface. Both the composition and the thickness of the passive film change continuously with potential5 and pH.4 A primary orienting idea about the electrochemical conditions for passivation can be obtained from a potential vs. pH (i.e., Pourbaix) diagram.12 Under anodic conditions tin passives over a range of pH values around neutrality, since the solubility of the various tin oxides goes through a minimum at a pH close to 8.5. This behavior is due to the amphoteric nature of the metal: solubility of tin hydroxides is higher at lower pH values, while at higher pH values the formation of stannites and stannates increases the solubility of the formed oxides.13 Regarding the composition of the film there is some disagreement in the literature1,14-29 and the electrochemical oxidation mechanism remains unclear.
With respect to the study of tin in slightly alkaline solutions, such as borate solutions, there is an extensive bibliography.14-20 Early studies suggested that the initial oxidation stages leading to passivity could be defined as a dissolution-precipitation mechanism of either Sn(OH)2 or SnO.21,22 Shah and Davies23 suggested two parallel processes, metal dissolution and oxide formation. Stirrup and Hampson1 reported, using a rotating disc technique in strongly alkaline solutions, that the first oxidation stage of tin is a dissolution-precipitation of SnO (or Sn(OH)2), while the second stage was a solid state process leading to the formation of a Sn(OH)4 film and finally permanent passivity results from a layer of SnO2 covering the tin metal. He also stated that this layer formation process would make the final passive film composite with a continuous film of SnO2 sandwiched between the tin metal and a noncontinuous film of SnO and/or Sn(OH)2. However, the XPS studies reported by Ansell et al.24 showed that the passive film consisted of SnO2 and Sn(OH)4, not detecting Sn(II) species. Thus, in general, it is proposed that the result of the first passivation step is a thin layer of Sn(IV) hydroxide. In very alkaline solutions, Shams El Din and Abd El Wahab25 concluded that the primary passivation of tin occurs when the metal is covered with a film of Sn(OH)2 or SnO, and then permanent passivity occurs with a continuous layer of Sn(OH)4 covering the surface, while Varsanyi et al.,14 using Mössbauer spectroscopy, found that the film formed at low potentials was duplex, consisting on highly amorphous Sn(OH)2 (or hydrated SnO) and SnO2 (or Sn(OH)4), while at higher potentials the passive layer consisted only of Sn(IV) hydroxide or oxide. The most stable species under anodic polarization is SnO2.H2O26 and, as dehydration of Sn(OH)4 to SnO2 is highly irreversible,26 it can be assumed that the dehydration to SnO2.H2O is also irreversible. Bianchi27 reported that oxygen is evolved on a SnO2.H2O film. A chemical transformation must then occur after the first passivation stage, prior to oxygen evolution reaction. Studies of the films formed during oxygen evolution done by Hampson and Spencer,28 using electron diffraction, showed them to be duplex, consisting of a thick, poorly adherent and generally amorphous layer of 5SnO.2H2O overlaying a thin, strongly adherent film of small crystals of SnO2.H2O.28,29 More recently, it has been reported that tin behaves as a typical valve metal and that the growth of the film involves field-assisted migration of ions through the oxide.20 The films were found to be nonstoichiometric, having an excess of tin atoms or oxygen vacancies that resulted in n-type conductivity.7
Previous contributions of our group presented new electrochemical addition methods of metals on SnO2 that can improve its gas-sensing behaviour.30,31 As the knowledge of the electronic properties of the semiconductor material and the influence in this properties of the metal addition are fundamental for these applications and given that electrochemistry can provide the ground for this understanding, we have started a characterization of tin oxides in borate buffer solution at pH 7.5 following previous work of our lab in the growth of iron oxide films under electrochemical control.32, 33 The results here presented include the reassignment of the oxidation/reduction voltammetric peaks and, although both techniques might lead to different films as, for example, cyclic voltammetry deals with transient conditions while impedance spectroscopy as here employed looks at stationary conditions, we try to give a brief sketch of the correlation of voltammetric with impedance data. Our results suggest the buildup of an energy barrier at the surface (located presumably at the oxide/solution interface) that strongly conditions the subsequent electrochemical behavior of the oxide layer (e.g. reduction potentials). Moreover, we report our first results by EC-STM of this system that allows to distinguish different electronic properties of the oxide layer formed at different anodic potentials. A sketch of the different behavior of tin when compared with iron32,33 is also presented.
Experimental
Polycrystalline tin samples used in this work have a purity of 99.999% (Goodfellow) and were cut as dishes with 10 mm of diameter and 1 mm of width. Prior to the electrochemical measurements, polycrystalline tin was mechanically polished to a mirror finished surface using an alumina paper with a grain size of 3 mm in order to have a proper roughness of the surface, as showed in Figure 1.
All results are expressed in terms of the projected geometrical area (1 cm2). The electrodes were mounted in a standard three-electrode cell, the auxiliary-electrode was a platinum grid and the reference electrode was a Ag/AgCl electrode from Metrohm.For the electrochemical studies, either a manual potentiostat (HQ instruments 105) connected to a signal generator (EG&G Universal Programmer 175) and an analog plotter (Philips PM 8033) or a digital electrochemistry equipment "SOLARTRON SI 1287 Electrochemical Interface" was used. It has a resolution of 1 mV in voltage and 1 pA in current, with maximum ranges of ± 14.5 V and ± 2 A respectively. It has an Electrochemistry/Corrosion software (CorrWare v1.2), (SOLARTRON, Farnborough, England). The capacitance measurements were performed with a frequency response analyzer (Solartron 1255).
An optical interferometric microscope was used for the study of the roughness of different samples. The equipment used was a ZYGO model GPIxp (Zygo Corporation, Connecticut), which can have a lateral resolution of 500 nm and an axial resolution of 0.1 nm, with a maximum range of 100 mm in Z.
Electrochemical scanning tunneling microscopy (ECSTM) studies were performed at room temperature using a Molecular Imaging microscope head (Phoenix, AZ, USA) controlled by a Nanoscope IIIa electronics (Digital Instruments Veeco Metrology Group, Sta. Barbara, California, USA). The STM electrochemical cell was made of Teflon and exposed a sample area of 0.42 cm2 to the solution through an O-ring. A Pt wire is used as an auxiliary electrode and a homemade true Ag/AgCl reference electrode with a double membrane (Modified Molecular Imaging (Phoenix, AZ, USA) Ag/AgCl electrode). STM tips were prepared by electrochemical etching of a Pt/Ir 80:20 wire in an acidic CaCl2 solution and covered with Apiezon wax. Tips prepared with this method present a large working potential range in our working electrolytic media.32 All the EC-STM images were recorded in the constant current mode and at constant applied sample potential. Typical tunneling setpoint currents range from 3 to 5 nA.
All the electrochemical experiments presented within this work were done in a boric/borate buffer solution at pH 7.5 and at 298 K. The solution was prepared using Milli-Q water and by mixing H3BO3 (p.a. grade, Merck) 0.35 molL-1 and Na2B4O7. 10H2O (p.a. grade, Merck) 0.0375 mol L-1. Prior to the electrochemical experiments, the solution was bubbled with Argon gas in order to remove the oxygen, but no bubbling was necessary during the experiments, that were performed under static hydrodynamic conditions.
Results
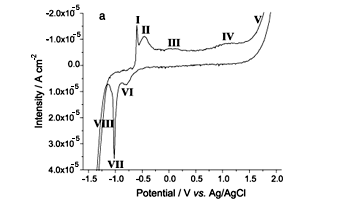
Figure 2 shows the electrochemical response of a polycrystalline tin polished sample immersed in a borate buffer solution at pH 7.5. Figure 2a describes the peaks observed in a cyclic voltammogram and associated to defined processes. The description is qualitatively similar to that previously reported at the same conditions.14,17 Only to say that in the first cycle the initial anodic branch may be influenced by the pre-treatment, mainly increasing the hydrogen evolution thus making less distinguishable the two fist oxidation peaks. For this reason, Figure 2a shows the second scan as more representative, which is identical to all subsequent scans. If we choose the potential limit of the cathodic scan as _1.6 V, hydrogen evolution is clearly present and then we have used this value as the lowest reduction potential in all samples. The anodic potential limit in Figure 2a at +2.1 V allows to show the passivation layer potential plateau prior to oxygen evolution. Four oxidation peaks (from I to IV) and two reduction peaks (VI and VII) are labeled in Figure. 2a, together with both oxygen and hydrogen evolution (labeled as V and VIII, respectively). The assignment of the peaks will be maintained throughout the paper in order to identify them.
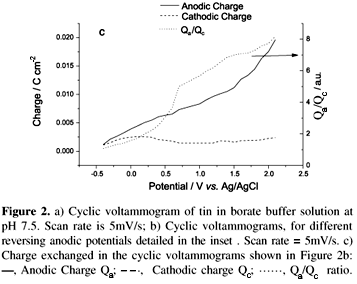
Kapusta17 early reported that the potential position of the reduction peak here assigned as VII shifted to more cathodic potentials when the scan anodic potential limit was shifted to more anodic values. Additionally, we observed that the shift is permanent, i.e., even if we stop 15 minutes at _1.6 V and we perform a new potential cycle turning back to the former anodic potential limit, peak VII does not regain its original position. In order to not include these history-dependent shifts, we have performed the following procedure: prior to each experiment, samples were reduced during 15 minutes at _1.6 V, and the voltammograms (Figure 2b) were done beginning the scans in the lowest anodic potential limit and then increasing it steeply just to reach +2.1V. Following this procedure of increasing the anodic potential limit, there is no significant change of both area and position of the oxidation peaks but indeed there is a change of both the position of the main reduction peak (that we always attribute to the same reduction process, the one labeled as peak VII) which is shifted to more negative values and the current intensity of HER, which is lowered (in absolute value). Moreover, it can be also observed that peak VI is only present when the anodic potential limit is higher than +1 V. The charge exchanged during the anodic and cathodic scans in the different cyclic voltammograms is also represented in Figure. 2c, together with the ratio between them.
With this behavior in mind, a systematic protocol was established in order to have a better insight on the causes that induce the observed changes: the tin samples were reduced during 15 min at _1.6 V, thereby stepped to the chosen anodic potential and there oxidized for 10 minutes. Afterwards, we linearly scan (at a scan rate of 5 mV/s) to _1.6 V. The study has been done at anodic potential limits ranging from _0.7 to 2.1 V in steps of 0.1 V, and the observed behavior is similar to that observed by cyclic voltammetry. The influence of diffusion processes or migration inside the film was also investigated using higher scan rates (i.e., 15 mV/s) and shorter oxidation times either 1 minute or 10 seconds. All the experiments show again similar behaviors.
As peak VII shifts to more negative potentials, we have performed a fitting for this peak. Figure 3 is showing the peak shift and the result of the fitting procedure. Additionally, and in order to characterize the shift in the HER wave, Figure 3 also represents the reduction current density at _1.59 V as a function of the applied anodic potential limit. One important fact in this figure is that when the sweep of the cyclic voltammetry has an anodic potential limit lower than, at least, -0.2V vs. Ag/AgCl, i.e., when only peaks I, II, VII, and VIII are present, the hydrogen evolution does not change.
EC-STM studies have been performed in this system following the procedure used for iron.32-34Figure 4 shows the in-situ EC-STM images of the surface after scanning up from _1.6 V to _0.5 V (Figure 4a) and of the surface after scanning up to 1.2 V (Figure 4b). Figure 4a shows that the first oxide formed on the surface are small nuclei while Figure 4b shows how at higher oxidation potentials the semiconducting properties of the oxide layer induce a loss of the STM image similar to what happens for iron anodic films.34
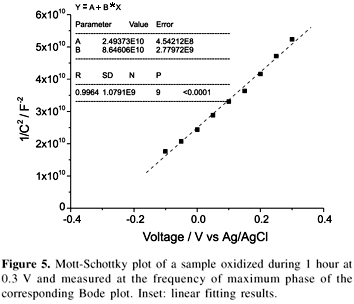
Impedance measurement analysis were made along the potential region in the oxidation plateau (from 0 to 1.2 V) as shown in the voltammetric study. Thus, after 1 h of oxidation at the chosen potential (the time was chosen in order to be long enough to have a full growth of the film, until oxidation current drops drastically (< 5 mA/cm2)), the Bode and Mott-Schottky plots were acquired. The Bode plots were qualitatively similar for all the oxidation potentials and times investigated, showing a unique phase maximum of more than 800 around 5 Hz. With respect to the Mott-Schottky plots, at low oxidation potentials a linear behavior is observed (Figure 5). The Mott-Schottky plot has been acquired by progressively reducing the applied potential. Thus, the oxide layer is formed during 1h at 0.3V and then we acquire the plot beginning at 0.3V and reducing the potential each 0.05V up to _0.1V, thereby avoiding changes of composition and thickness that would result of plot acquisition by increasing the potential. At higher oxidation potentials the plot changes the shape but giving in any case a n-type semiconducting behavior. With respect to the interpretation of these results, it is generally agreed that surface or bulk states are present in the bandgap of anodically formed semiconductor electrodes.5 When these states give rise to a degenerated semiconducting oxide layer (for example, by inducing a high ND), the Mott-Schottky plots are no longer linear and it is for this reason that the interpretation of semiconducting properties of the passive film using the Mott-Schottky approach is still a matter of discussion.5
Discussion
The results presented above state that the initial oxidation of tin takes place in two overlapped peaks (I and II). These two peaks have been attributed before17 to two differentiated oxidation processes corresponding to the oxidation of metallic tin to Sn(II) (peak I) and of Sn(II) to Sn(OH)4 (peak II), thereby attributing the unique reduction peak (peak VII) observed when the scan is reversed at a less positive potential than peak III appears, to the reduction of both species to metallic tin. The presence of a unique reduction peak, might be indicative of a single reduction process, so we propose that in the first two oxidation peaks metallic tin is only oxidized to Sn(II), probably beginning with the hydroxylation of the metallic tin surface (peak I), afterwards forming tin (II) hydroxide by a dissolution-precipitation mechanism (peak II) since Sn(II) species have a low solubility in the non-complexant borate buffer solutions (Kps (Sn(OH)2)= 5.45x10-27). Thus, peak VII will correspond to the reduction of Sn(II) to metallic tin. It must be pointed out that, if the sweep of the cyclic voltammetry has an anodic reversing potential less positive than -0.2 V, the oxidation-reduction process is completely reversible because the initial surface is recovered after an oxidation/reduction cycle, as observed by in-situ EC-STM. In this case, hydrogen evolution does not change confirming the recovering of the surface by metallic tin. Moreover, in-situ EC-STM has detected the oxidation of tin to begin at potentials where only peak I is present and, as discussed in the Results section, the influence of the possible oxidation of atomic hydrogen introduced into the tin lattice gives to two poorly distinguishable peaks in the first cyclic voltammograms, thereby discarding peak I to be connected with HER. In the second and subsequent cyclic voltammograms, hydrogen incorporation is less pronounced and peak I does not change. Charge measurements given in Figure 2c also give support to the assignment of peaks I/II to the formation of Sn(II) because until potentials around _0.1 V the ratio between the anodic and the cathodic charge is close to 1, showing again that the oxido-reduction process is almost fully reversible and that peak I could not be associated with hydrogen oxidation but with oxidation of tin. In conclusion, the first oxidation-reduction cycle (peaks I, II, VII) of tin is chemically similar to that reported in the case of iron32, just obtaining a quasi-metal or p-doped semiconductor allowing to incorporate electrons from the tip to the film. The main difference is that, in the case of iron, the electrolysis at a equivalent potential range gives a thicker film and a higher amount of anodic charge transfer mainly because the Kps of iron (II) hydroxide is twelve orders of magnitude higher than in the case of tin, and then a faster passivation process takes place.32
Scanning up in the anodic branch up to 0 V, oxidation peak III appears. To our knowledge, this peak was only reported and commented once17 and it was attributed to the electrochemical dissolution of the previously formed oxide. Although with our experimental evidences it is still difficult to assign the peak to a definite process, the experimental changes in the electrochemical response detected at more anodic potentials than peak III appears, point to a different interpretation. The observed decrease in the electrocatalytic activation of hydrogen evolution and the shift of peak VII position to more cathodic potentials (Figure 3) indicate the presence of a new oxide and the formation of hydrated tin (IV) oxide would begin. All these effects are probably partially kinetic, but also a different electronic response have to be taken into account, because oxidizing at these potentials the Mott-Schottky plots exhibit a linear behavior typical of an n-type semiconductor oxide (Figure 5). Thus,the energy barrier created by the semiconducting layer at the electrode surface35 can explain the shift of peak VII. In the negative sweep of the voltammogram, free electrons in the CB must overcome this barrier in order to reduce the oxide. Likely, as the positive potential limit is increased between peak III and peak IV, the barrier would increase in height due to a shift of the CB edge away from the Fermi level, resulting in the progressive oxidation from Sn(II) to Sn(IV) (lower concentration of "oxygen vacancies" or Sn(II) sites, which act as donor dopant atoms, like in other semiconducting oxides, as Fe(II) in iron oxides36). Thus it appears that in this case, Sn(II) progressively oxidizes to form a hydrated tin(IV) oxide SnOx (where 1<x<2). This picture is in agreement with the loss of the EC-STM image when oxidizing at higher oxidation potentials (Figure 4b), as the formation of a more stoichiometric tin (IV) oxide would shift the CB edge potential away from the range of potentials available to the fixed potential applied to the tip. Unreduced tin (IV) oxide will be the responsible for the decrease of hydrogen evolution current (Figure 3). The presence of an unreduced oxide on the metallic tin surface is also supported by the high increase of the anodic/cathodic charge ratio (Figure 2c), with is a measure of the irreversibility of the oxidation/reduction process, and this behavior contrasts with what happens in the oxidation/reduction processes of iron, where the formation of Fe (III) oxides gives rise to additional reduction peaks and the overall oxidation/reduction process is completely reversible when accessible negative reduction potentials are reached.32
At far anodic potentials, over 0.9 V, peak IV appears. Although at the present stage of the study we cannot unambiguously attribute this peak, the formation of a more stoichiometric hydrated tin (IV) oxide will only be attained at these potentials thereby giving rise to more complex Mott-Schottky plots, that have been attributed to the presence of surface or bulk states in the bandgap of the anodically formed semiconductor electrodes,5 in contrast to the case of iron where only one linear Mott-Schottky region, also attributed to the n-type semiconducting behavior of Fe(III) oxides, is present.32
For anodizations in the potential region ranging between 1.3 and 2.1 V, no further shift of peak VII is observed. Contrarily, only after oxidizing at potentials higher than 1 V peak VI is well defined and, moreover, a further decrease of hydrogen evolution is observed. All these processes might indicate the thickness growth of the tin (IV) oxide film, together with a change of its crystalline properties, that could be affected by the oxygen evolution. Peak VI is attributed to the reduction of tin (IV) to tin (II). This process is not complete since, as previously discussed, there is always a certain amount of tin (IV) oxide that is not reduced. The anodic/cathodic charge ratio increase to values much greater than 1 when increasing the oxidation potential (see Figure 2c) is a clear evidence of the existence of an irreversible oxidation/reduction process that would give rise to a remaining oxide layer that would accumulate in successive cycles. Regarding the oxygen evolution current (labeled as V), the intensity decreases with subsequent cycles, thereby confirming that the passive film is somehow irreversible and that the remaining oxide accumulates from cycle to cycle and passivates efficiently the tin surface. The main features of the hydrogen evolution (labeled as VIII) are fully connected to the kind of surface existing at the end of the cathodic reduction, that is, metallic tin, non-crystalline doped oxide and crystalline doped oxide.
Conclusions
The electrochemical oxidation and reduction processes of a tin polycrystalline surface in borate buffer solutions at pH 7.5 have been systematically investigated. The results have allowed us to discuss the different formed oxides with respect to the anodization potential and the reassignment of some peaks and plausible arguments for the definition of the peaks in the oxidation plateau have been presented. The shift of the major reduction peak when the anodization potential is increased has been attributed to the variation of the doping concentration in the formed oxides. Moreover, the formation at high oxidation potentials of a tin (IV) oxide, not reduced even stopping at the HER region, is responsible for hindering the hydrogen evolution. Systematic impedance studies for the obtention of quantitative values of the electronic properties of the passive film are currently under development in order to compare them with new EC-STM results.
Received: January 15, 2003
Published on the web: August 8, 2003
- 1. Stirrup, B. N.; Hampson, N. A.; J. Electroanal. Chem. 1976, 67, 45.
- 2. Young, L.; Anodic Oxide Films; Academic Press: New York, 1961.
- 3. Vijh, A. K. In Anodic Oxide Films: Influence of Solid State Properties on Electrochemical Behavior of Oxides and Oxide Films; Diggle, J. W., ed.; Marcel Dekker: New York, 1973, vol. 2.
- 4. Seruga, M.; Metikos-Hukovic, M.; J. Electroanal. Chem. 1992, 334, 223.
- 5. Metikos-Hukovic, M.; Omanovic, S.; Jukic, A.; Electrochimica Acta 1999, 45, 977.
- 6. Seruga, M.; Metikos-Hukovic, M.; Valla, T.; Milun, M.; Hoffschultz, H.; Wandelt, K.; J. Electroanal. Chem. 1996, 407, 83.
- 7. Burleigh, T. D.; Gerischer, H.; J. Electrochem. Soc. 1988, 135, 2938.
- 8. Giannetti, B. F.; Sumodjo, P. T. A.; Rabockai, T.; Souza, A. M.; Barboza, J.; Electrochimica Acta 1992, 37, 143.
- 9. Bojinov, M.; Salmi, K.; Sundholm, G.; J. Electroanal. Chem. 1993, 358, 177.
- 10. Giannetti, B. F.; Sumodjo, P. T. A.; Rabockai, T.; J. Appl. Electrochem. 1990, 20, 672.
- 11. Drogowska, M.; Ménard, H.; Brossard, L.; J. Appl. Electrochem. 1991, 21, 84.
- 12. Pourbaix, M.; Atlas of Electrochemical Equilibria in Aqueous Solutions; Pergamon Press: Oxford, 1966.
- 13. Bailar, J. C.; Eméleus, H. J.; Nyholm, R.; Trotman-Dickenson, A. F.; Comprehensive Inorganic Chemistry, Pergamon Press: Oxford, 1973.
- 14. Varsanyi, M. L.; Jaen, J.; Vertes, A.; Kiss, L.; Electrochim. Acta 1985, 30, 529.
- 15. Kapusta, S.; Hackerman, N.; Electrochim. Acta 1980, 25, 949.
- 16. Kapusta, S.; Hackerman, N.; Electrochim. Acta 1980, 25, 1001.
- 17. Kapusta, S.; Hackerman, N.; Electrochim. Acta 1980, 25, 1625.
- 18. Kapusta, S.; Hackerman, N.; J. Electrochem. Soc. 1981, 128, 327.
- 19. Kapusta, S.; Hackerman, N.; J. Electrochem. Soc. 1982, 129, 1886.
- 20. Metikos-Hukovic, M.; Resetic, A.; Gvozdic, V.; Electrochim. Acta 1995, 40, 1777.
- 21. Goldschmidt, H.; Eckardt, G.; Z. Phys. Chem. 1906, 56, 385.
- 22. Foerster, F.; Dolch, M.; Z. Elektrochem. 1909, 16, 599.
- 23. Shah, S. N.; Davies, D. E.; Electrochim. Acta 1963, 8, 703.
- 24. Ansell, R. O.; Dickinson, T.; Povey, A. F.; Sherwood, P. M. A.; J. Electrochem. Soc. 1977, 124, 1360.
- 25. Shams El Dim, A. M.; Abd El Wahab, F. M.; Electrochim. Acta 1964, 9, 883.
- 26. Sillen, L. G.; J. Chem. Educ. 1952, 29, 600.
- 27. Bianchi, G.; Chimica Ind. Milano 1947, 29, 295.
- 28. Hampson, N. A.; Spencer, N. E.; Br. Corros. J. 1968, 3, 1.
- 29. Pugh, R.; Gabe, D. R.; Warner, M.; Corros. Sci. 1967, 7, 807.
- 30. Díaz, R.; Arbiol, J.; Cirera, A.; Sanz, F.; Peiró, F.; Cornet, A.; Morante, J.R.; Chem. Mater. 2001, 13, 4362.
- 31. Díaz, R.; Arbiol, J.; Sanz, F.; Cornet, A.; Morante, J.R.; Chem. Mater. 2002, 14, 3277.
- 32. Díez-Pérez, I.; Gorostiza, P.; Sanz, F.; Müller, C.; J. Electrochem Soc. 2001, 148, B307.
- 33. Díez-Pérez, I.; Gorostiza, P.; Sanz, F.; Müller, C.; Proceedings of the Symposium "Scanning Probe Microscopy for Electrode Characterization and Nanometer Scale Modification", The Electrochemical Society Inc.: Pennington NJ, 2001, vol. 2000-35, p. 122.
- 34. Díez-Pérez, I.; Gorostiza, P.; Sanz, F.; J. Electrochem Soc. 2003, 150, B348.
- 35. Morrison, S.R.; Electrochemistry at Semiconductor and Oxidised Metal Electrodes; Plenum Press: New York, 1980.
- 36. Büchler, M.; Schmuki, P.; Böhni, H.; Stenberg, T.; Mäntylä, T.; J. Electrochem. Soc. 1998,145, 378.
Publication Dates
-
Publication in this collection
15 Sept 2003 -
Date of issue
Aug 2003
History
-
Received
15 Jan 2003 -
Accepted
08 Aug 2003