Abstracts
Glycyrrhizinic acid (GL), also known as glycyrrhizin, is a triterpene saponin, a natural product found on the root of Glycyrrhyza glabra L. ("licquorice"), used worldwide as sweetener and in the traditional eastern medicines. This review is focused on a series of new derivatives synthesized using GL and its aglycon, glycyrrhetinic acid (GLA), as starting materials, the pharmacological activities described for those compounds, as well as new activities reported for GL and GLA themselves.
glycyrrhizin; glycyrrhetic acid; glycyrrhetinic acid; licquorice; organic synthesis; pharmacological activity
O ácido glicirrizínico (GL), também conhecido como glicirrizina, é uma saponina triterpênica, um produto natural encontrado na raiz de Glycyrrhyza glabra L. ("licquorice" ou "alcaçuz"), utilizada mundialmente como edulcorante e também na medicina tradicional do Oriente. Este artigo de revisão enfoca os novos compostos sintetizados usando GL ou sua aglicona, o ácido glicirretínico (GLA), como materiais de partida e as atividades farmacológicas descritas para os mesmos e seus derivados.
REVIEW
Glycyrrhizin and glycyrrhetic acid: scaffolds to promising new pharmacologically active compounds
Cedric S. GraebinI,II; Hugo VerliI; Jorge A. GuimarãesI,* * e-mail: guimar@cbiot.ufrgs.br
ICentro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Avenida Bento Gonçalves 9500, 91500-970 Porto Alegre-RS, Brazil
IIDepartamento de Química, Instituto de Ciências Exatas, Universidade Federal Rural do Rio de Janeiro, BR 465, km 7, Bairro Cidade Universitária, 23890-000 Seropédica-RJ, Brazil
ABSTRACT
Glycyrrhizinic acid (GL), also known as glycyrrhizin, is a triterpene saponin, a natural product found on the root of Glycyrrhyza glabra L. ("licquorice"), used worldwide as sweetener and in the traditional eastern medicines. This review is focused on a series of new derivatives synthesized using GL and its aglycon, glycyrrhetinic acid (GLA), as starting materials, the pharmacological activities described for those compounds, as well as new activities reported for GL and GLA themselves.
Keywords: glycyrrhizin, glycyrrhetic acid, glycyrrhetinic acid, licquorice, organic synthesis, pharmacological activity
RESUMO
O ácido glicirrizínico (GL), também conhecido como glicirrizina, é uma saponina triterpênica, um produto natural encontrado na raiz de Glycyrrhyza glabra L. ("licquorice" ou "alcaçuz"), utilizada mundialmente como edulcorante e também na medicina tradicional do Oriente. Este artigo de revisão enfoca os novos compostos sintetizados usando GL ou sua aglicona, o ácido glicirretínico (GLA), como materiais de partida e as atividades farmacológicas descritas para os mesmos e seus derivados.
1. Introduction
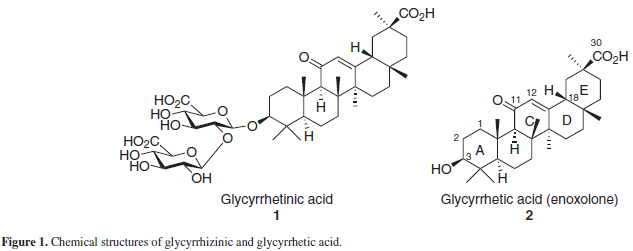
Glycyrrhizinic acid or glycyrrhizin (GL) (1), Figure 1 is a triterpene saponin found in the root of the Leguminosae Glycyrryza glabra L., commonly known as "licquorice" or 'licorice' and is used worldwide as a natural sweetener and flavoring additive,1 as well as in the tradicional Chinese, Indian and Tibetan medicines.2 GL, along with its aglycon, glycyrrhetic or glycyrrhetinic acid (GLA), also known as "enoxolone" (2), are the most studied and abundant compounds from this plant and are commercially obtained from the crude extract of the licquorice root. As a sweetener, GL is reported to be 30-50 times sweeter than sucrose.1-3
GL, its aglycon and liquorice extracts are known for its wide range of biological activities, such as anti-inflamatory, anti-allergic, anti-arthritic, antioxidant, hypolipidaemic, antiviral and antitumoral, as well as presenting hepato and imunoprotective effects in humans and animals.3,4 Both GL and its aglycon have been employed in the past decades as starting materials for the chemical synthesis of new derivatives, many of them with interesting biological activities. Some reviews have been published reporting new GL and GLA derivatives, as well as their activities.1,3 The current review is focused, therefore, in recent data on the syntheses and the synthetic strategies employed for GL and GLA derivatives, published from 2003 onwards, the pharmacological activities of these synthesized derivatives, and new pharmacological activities reported for both GL and GLA.
2. Chemical Modifications of GL
2.1. Immunomodulating derivatives
Guided by previous results indicating that GL-aminoacid or GL-peptide conjugates can exert immunomodulating activities,3,5 Kondratenko et al.6 have prepared some GL derivatives containing residues of L-glutamic acid and its α-,γ- and α,γ-diesters (4-14, Scheme 1) using 1 or its 30-methyl ester 3 as starting materials, employing dicyclohexylcarbodiimide (DCC) as coupling promotor. The deprotected derivatives 5, 7 and 14 were obtained from selective hydrogenolysis of the protected esters 4, 6 and 13, respectively, with Pd/C and 12 was obtained with removal of the tertbutyl ester group with trifluoroacetic acid TFA. Compounds 5 and 12, when administered intraperitonially in mice, had opposite effects in the production of antibody-forming cells (AFC), when compared with control (isotonic saline solution) animals. Compound 5 increased the number of AFCs, while 12 lowered the AFC count, indicating that 5 may exert an immunostmiulating effect, while 12 may act as an immunosupressor. Although only these two compounds were tested in this assay, a brief comparison of 5 (a compound with its three carboxylic groups esterified with glutamic acid methyl ester) and 12 (esterified only at the glycosyl carboxylic groups) points to the importance of modifications at the C-30 carboxylic group to this activity. Moreover, compound 13, when orally administered in mice, increased serum levels of hemagglutinin when compared with control animals, indicating that 13 can exert an immunostimulating activity.
On the search of new immunomodulating GA-aminoacid conjugates, the same research group synthesized GL-cysteine derivatives 15-22 (Scheme 2) employing DCC/N-hydroxysuccinimide (HOSu) or Woodward's reagent K as coupling agents. Chemoselectivity for the glycosidic carboxylic acids was achieved by performing the DCC coupling at 0-5 ºC. At 20-22 ºC, the three carboxylic acid groups of GL were coupled. Compounds 18 and 22, administered intraperitonially, presented an imunostimulating activity in mice, increasing the number of antibody-producing cells when compared to control (isotonic saline solution) animals.7 Another work from the same group yielded GL-aminoacid ester derivatives 22-32 (Scheme 3) in a similar fashion. Compounds 23, 26 and 29, administered intraperitonially, presented in vivo immunomodulating activity, increasing the serum level of agglutinins and hemolysins when compared to control (isotonic saline solution) animals.8 Although other compounds in both series were not tested, the chemical modifications performed may indicate that the esterification of glucosyl carboxylic groups of GL with aminoacid derivatives such as Cyz(Bz)OH, Cyz(Bz)-Val(OH), glycine ethyl ester, alanine butyl ester or valine butyl ester, with the C-30 carboxyl group either unmodified or esterified with methyl groups lead to immunostimulating compounds. In one case, esterification of the three carboxyl groups of GL with glutamic acid methyl ester lead to an immunostimulating compound (5), whereas the derivative with only the glucosyl acid groups esterified (12) lead to an immunosupressor compound, which indicates that the carboxyl group of this aminoacid derivative is important for the interaction of the compound with its molecular target.
2.2. Antiviral derivatives
As previously mentioned, GL was reported as an anti-viral compound, being moderately active against HIV and herpes simplex virus.3,4 Exploring the chemical versatility of this saponin, Baltina Jr. et al.9 employed the technique described in Schemes 1-3 to obtain GL-lysine conjugate compounds 33-39 (Scheme 4). Compound 39 was tested against HIV strains in MT-4 cell culture assay and showed moderate anti-HIV activity (IC50 = 0.145 µM), somewhat greater than its precursor GL (IC50 = 0.304 µM).
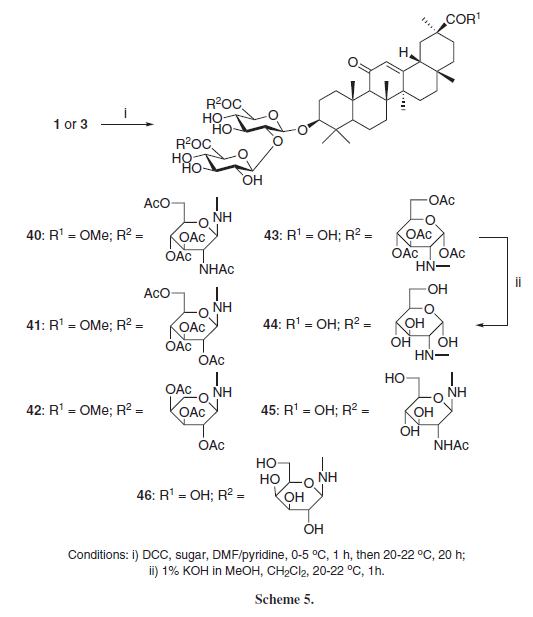
Kondratenko et al.10 synthesized GL-aminosugar amide conjugates 40-46 employing DCC as coupling agent and some aminosugars selectively at the glucoside carboxyl groups (Scheme 5). Compound 45 presented in vitro antiviral activity against herpes simplex virus 1 (HSV-1) (IC50 = 0.0032 µM) and compounds 45 and 46 presented moderate in vitro antiviral activity against HIV-1 (IC50 = 0.35 and 0.09 µM, respectively), lower than the standard drug Azidothymidine (AZT) (IC50 = 0.014 µM), suggesting that the presence of more polar hydroxyl groups in 46, instead of the N-acetyl group from 45, is important for the anti-HIV activity of these derivatives.
Considering SARS-Coronavirus, Hoever et al.11 synthesized and tested the derivatives 18, 45, as well as GL derivatives 47-59 and GLA derivatives 60 and 61 (Figure 2). Glycoderivatives 55-58 presented the higher in vitro antiviral activitiy against this virus (IC50 = 8 ± 2, 50 ± 10, 5 ± 3 and 16 ± 1 µM, respectively), greater than GL itself (365 ± 12 µM). Derivatives 47, 48 and 49 were also active (IC50 = 40 ± 13, 35 ± 7 and 139 ± 20 µM), while 57-61 were inactive (IC50 > 1000 µM), indicating that, for aminoacid-GL conjugates, the free 30-COOH group is important for this activity. Carbohydrate-free compounds, such as 2, 60 and 61, were also inactive (IC50 > 1000 µM), indicating that the glycosyl moiety is important to the antiviral activity of the derivatives, probably because Coronaviruses are highly glycosylated, especially in the spike proteins, that project from the surface of the viral envelope. So, according to the authors, the glycosyl moiety in the active derivatives is important for interactions with these and other glycosylated proteins in the viral surface. Compounds 55-58 were also found to be cytotoxic against VERO cell cultures.
2.3. Anti-allergic derivatives
Employing Streptococcus LJ-22 bacteria as biocatalysts, Park et al.12 synthesized the glycyrrhetinic acid-3-O-β-D-glucoronide (GAMG, 62). This monoglucoronide presented greater in vitro anti-allergic activity (measured by the inhibition of β-hexosaminidase release from RLB-2H3 cells) than GL and comparable with GLA (IC50 = 0.28, 0.37 and 0.23 nM, respectively), although the molecular basis for this action was not further explored. It seems that, although the authors considered that the three compounds presented anti-allergic activities, the carbohydrate portion of the molecule isn't essential for this activity, paving the way for new anti-allergic derivatives with simpler molecular patterns.
2.4. Anti-ulcer derivatives
Alternatively, the obtaining of glycoderivates of GL was also accessed for anti-ulcer activity. Starting from penta-acetyl GL (63) or penta-acetyl GL 30-methyl ester (64), Kondratenko et al.13 synthesized new saponin compounds with monossacharide residues of glucose and galactose through ester bonds (65-69, Scheme 6). Compound 69, in the model of experimental rat stomach lesions induced by acetylsalicylic acid, expressed anti-ulcer effects at the dose of 25 mg kg-1 (orally administered), against 100 mg kg-1 of the standard drug carbenxolone or 50 mg kg-1 of GL. According to Williams et al.,4 in their evaluation of the pharmacological data associated with the use of GL as a food additive, GL is poorly absorbed orally, being metabolized in the gastrointestinal tract (by bacterial β-glucoronidases) to GLA (2) and in a lesser extent to GAMG (62), which is further metabolized to GLA. It is possible, therefore, that the greater anti-ulcer effect of those derivatives (when comparing their activities against GLA and GAMG) are a combination of the intrinsic anti-ulcer activity of the same along with a prodrug effect related to the probable metabolite GLA.
Such gastroprotective effects of GL derivatives (1, 2 and the per-acetylated GAMG (62) derivative 70) was also observed by Krause et al.14 employing as a model 29 strains of the bacteria Helicobacter pylori, known to contribute to a wide spectrum of gastroduodenal diseases, such as gastric and duodenal ulcers. Therefore, compound 2 was found as the most potent compound in the assay (MIC50/90 = 50/100 mg mL-1), followed by 70 (MIC50/90 ;= 400/ > 400 mg mL-1) and GL (MIC50/90 > 400 mg mL-1). According to the authors, this anti-H. pylori activity accounts for the anti-ulcer effect of 1 and 2.
3. Chemical Modifications of GLA 2
3.1. Antiviral derivatives
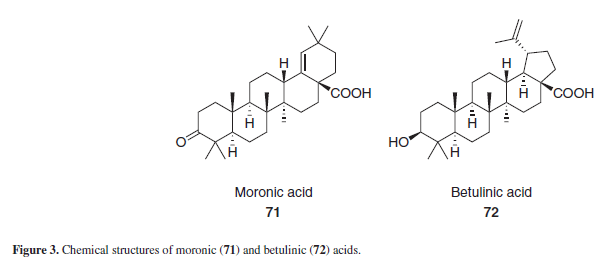
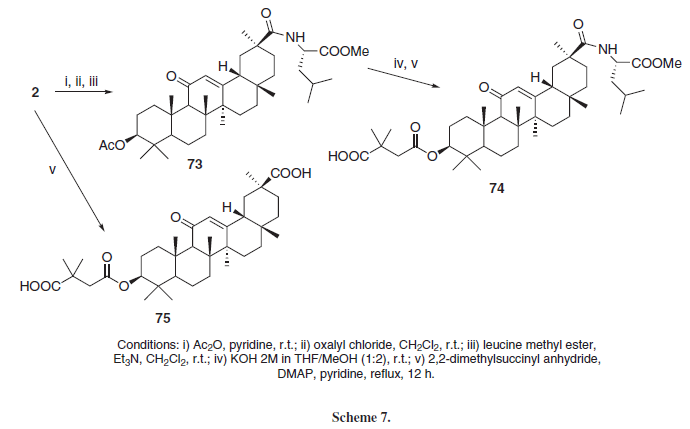
Moronic and betulinic acid (71 and 72), Figure 3, are naturally-ocurring triterpenoids, known for its antiviral properties, specially against HIV and herpes simplex virus. Some derivatives of both acids have anti-HIV activities as well.15 Guided by these active betulinic and moronic acid derivatives, Yu et al.15 synthesized two derivatives from GLA, 74 and 75 (Scheme 7), along with other triterpene aglycons. Those two compounds, when tested against in vitro HIV strains, showed no antiviral activity. According to Hoever et al.,11 the glycosyl moiety present in GL seems to be important for its interaction with highly glycosylated viral surface proteins, and GLA derivatives without glycosyl substituents exerted no antiviral activity against coronaviruses. Based on such proposal, it seems that the absence of glycosyl substituents in the GLA derivatives 74 and 75 may be responsible for its lack of anti-HIV activity as well.
3.2. Cytotoxic and antiproliferative derivatives
Chintharlapalli et al.16 inspired by some reports of cytotoxic activity of GLA and some oleane-related triterpenes that exerted its cytotoxic activity by acting as peroxisome proliferator-activated γ-receptor (PPAR-γ) agonists, synthesized 1,12-dienes (77-80, Scheme 8) from 3-keto-18β-GLA methyl ester (76) and 3-keto-18α-GLA methyl ester (76'). Only cyanoderivatives 79 and 80 presented in vitro activity as PPAR-γ agonists, leading the researchers to conclude that both the 30-methyl ester and the cyano group are essential for this activity, and that the stereochemistry at the C-18 position, although responsible for conformational differences between the 79 and 80, is not important for the activity.
In order to improve the cytotoxic activity reported for GLA through increasing of its lypophilic profiles, a series of oximes (82-91) were synthesized from 3-keto GLA (81) by Liu et al.17 (Scheme 9). Some of the compounds exhibited in vitro cytotoxic and apoptosis induction activity against HL-60 cells. The data presented by the authors indicates that the replacement of the hydroxyl group of 2 with carbonyl or oxime groups does not influence the antiproliferative activity, but compounds with alkoximino groups at C-3 and free C-30 acid groups presented greater antiproliferative activity and apoptosis induction. Moreover, C-30 esterified compounds with alkylated C-3 oxime groups lack apoptosis induction activity, while retain the antiproliferative activity, probably because the cytotoxic and the apoptosis induction effects are being modulated through different pathways.
Previous studies with some oleane-derivated triterpenes, such as the oleanoic acid derivatives CDDO and its methyl ester CDDO-Me18 indicated that 1-en-3-one derivatives with electronegative 2-substituent groups are highly cytotoxic, and the same effect was observed with GLA derivatives, such as 79 (Scheme 8).16 Based on such pharmacophoric group, Chadalapaka et al.19 synthesized analogs of 77 (79 and 92-95, Scheme 10) and other analogues with rearrangements in the C ring of GLA (99-101, Scheme 11), inserting along with the cyano group other electronegative groups. The cyano derivatives (with or without C-ring rearrangements) and the 2-trifluoro derivative 95 were the most active in in vitro anti-proliferative assays against four cancer cell lines (253JB-V and KU7 bladder cancer cells and Panc-1 and Panc-28 pancreatic cancer cells). These results seem to agree with the findings of Chintharlapalli et al.,16 where the cyano groups at C-2 were also important for the antiproliferative activity. It seems that, along with the electronegativity required at C-2, the size of this substituent group has a steric limit for a good interaction with the target receptor, which could explain the reason why more bulky substituents such as the methanesulfonyl and dimethylphosphonyl groups were inactive.
Paclitaxel (102, Scheme 13) is a natural product isolated originally from the barks of the Pacific Yew tree Taxus brevifolia, and is used in cancer therapy acting as a mitotic inhibitor. GLA (2) is also reported as presenting antitumor activity,3,4 and the conjugation of two antitumor agents with different mechanisms of action is reported to possibly increase the potency of both compounds and/or reduce its side effects and drug-resistance development.20 Guided by previous reports of the antitumoral activity of conjugates such as paclitaxel-camptothecin, paclitaxel-epipodophyllotoxin and camptothecin-epipodophyllotoxin, Nakagawa-Goto et al.20 synthesized new hybrid paclitaxel-conjugated compounds with several diverse scaffolds, including two amide derivatives coupled with GLA, 105 and 106 (Scheme 12). Those two compounds presented moderate to weak cytotoxic acidities against 6 cancer cell lines (105: IC50 ranging from 45 to 103 nM; 106: IC50 ranging from 147 to 652 µM) and, although less active than paclitaxel itself (IC50 ranging from 1.0 to 55 nM), they can serve as prototypes for new hybrid compounds with other active antitumoral molecules.
The same research group also studied the dehydrozingerone (DZ, 107, Scheme 13), a phenolic natural product reported as presenting anti-oxidant, anti-inflammatory and anti-tumor properties. GLA (2) was coupled with DZ derivatives (Scheme 14) leading to eleven GLA-DZ conjugates 108-118. Compounds 108, 109 and 110 presented in vitro cytotoxical effects against some cancer cell lines, being either GLA or DZ alone inactive at those assays.21 The researchers found that replacing methoxy (108-110) with ethoxy (112 and 113) or fluor (114) substituents diminished the cytotoxic activity of derivatives, while replacing the methyl (R2) groups with phenyl groups in compounds 115-118 abolished the activity.
3.3. Anti-inflammatory derivatives
Searching for in vitro anti-inflammatory and anti-oxidant activities, Maitraie et al.22 synthesized two 7-member lactones (119 and 120) from the Bayer-Villiger oxidation of the 3-keto derivatives 81 and 76 respectively, starting with 2 or the methyl ester 98. Both oxidation protocols did not affect the double bond at C-12 (Scheme 14). From these lactones, using two different synthetic pathways, they were able to obtain a series of derivatives (126-129 and 133-135). Compounds 120, 121 and 134 inhibited superoxide anion generation in rat neutrophils, while compounds 98, 124, 127, 129, 133 and 135 inhibited tumor necrosis factor-α (TNF-α) formation in RAW 264.7 cells in response to lipopolysaccharide (LPS). In this series of compounds, lipophilic group substituted at C-3 or C-30 and/or the presence of a carbonyl group at the ring A enhanced the anti-inflammatory activity.
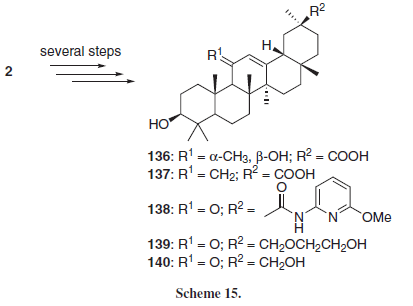
One of the most studied mechanisms of action of GL and GLA is their anti-inflammatory activity, attributed mostly to the inhibition of 11β-hydroxysteroid dehydrogenase (11β-HSD), an enzyme responsible for the metabolism of the anti-inflammatory glucocorticoid hormones cortisol (hydrocortisone) and its inactive form cortisone. The variant 11β-HSD-1 converts cortisone into cortisol, while the variant 11β-HSD-2 does the opposite, converting cortisol into cortisone, inactivating this hormone. GL and GLA are known to inhibit primarily 11β-HSD-2, presenting an indirect anti-inflammatory effect by increasing the levels of cortisol.2-4 In this context, Su et al.23 employing 2 as starting material for several chemical modifications, obtained a series of 11- and 30- modified GLA derivatives (136-140, Scheme 15). Compound 136 presented the higher rat 11β-HSD inhibitory activity of the series. Also, the authors found that the introduction of the 11α-methyl-11β-hydroxyl induces selectivity to the isoform 1 of the enzyme, which can be related to the role of HSD isoform 1 in converting the 11-hydroxyl group of cortisol to the 11-ketone group of cortisone. Compound 137 was the less active of the series, indicating that the presence of an oxygen atom (either as an alcohol or ketone), at C-11 position is important for the interaction with the rat 11β-HSD enzyme, probably because the C-11 oxo group is also present at the enzyme substrate cortisol. On the other hand, compound 139 also presented selectivity to the isoform 1 of the enzyme, indicating that a larger side chain at C-30 can be detrimental to the interaction of the compound with the rat 11β-HSD-2 enzyme.
Vicker et al.24 synthesized 7 GLA-derived amides via the DCC-promoted coupling (141-147, Scheme 16). Amide 142 inhibited (in vitro) the human 11β-HSD-2 enzyme, presenting little activity against the 11β-HSD-1 variant. The side chain of 142 was designed to mimic the side chain of cortisol/cortisone, while the keto group of the glucocorticoid hormone was replaced by the bioisostere amide group.
3.4. Proteasome inhibitors
Based on previous results indicating that some betulinic acid derivatives regulated the activity of the human 20S proteasome-ubiquitin complex, important in cell-stress responses and cell cycle regulations, Huang et al.25 synthesized a series of C-3 ester derivatives employing anhydrides or dicarboxylic acids (Scheme 17) and some amides employing aminoacids (Scheme 18). Compounds 162 (IC50 = 0.22 µM), 153 (IC50 = 0.29 µM), 163 (IC50 = 0.33 µM), 157 (IC50 = 0.35 µM) and 75 (IC50 = 0.87 µM) were the most active of the series, inhibiting in vitro the chemotrypsin-like activity of the human 20S proteasome complex with more potency than GLA (2, IC50 = 22.3 µM). The researchers concluded that free carboxylate groups seemed to increase the activity of the compounds, although not required for the compounds to present this activity (156 has an IC50 = 1.92 µM and 154 and 155 were inactive in this assay, with IC50 > 40 µM). Additionally, the presence of an aromatic ring and the position of the carboxylate group in that ring do not appear to be relevant for this activity (comparing the aromatic derivative 162, the aliphatic derivative 153 and the unsaturated derivative 157). Further modifications at C-30 appeared to diminish the proteasome activity.
3.5. Gene silencers
Short hairpin RNA (shRNA) are modified small interfering RNA (siRNA) molecules, capable of suppress gene expression of targeted genes with better properties (stability, cellular uptake and target specificity) than siRNA. Bang and Kim, starting from 2, synthesized the 11-desoxo-3,30-diol 166 which was selectively protected leading to 167. This protected diol was used as a hairpin loop for two shRNAs, 168 and 169, that acted as gene silencers against the EGFP gene, a gene that codifies the Green Fluorescent Protein (GFP) and is used as a template to measure the activity of siRNAs (Scheme 19), indicating that the GLA derivative 166 can act as a hairpin loop for the syntheses of other shRNAs targeting different and clinically relevant genes.26
3.6. Antioxidant derivatives
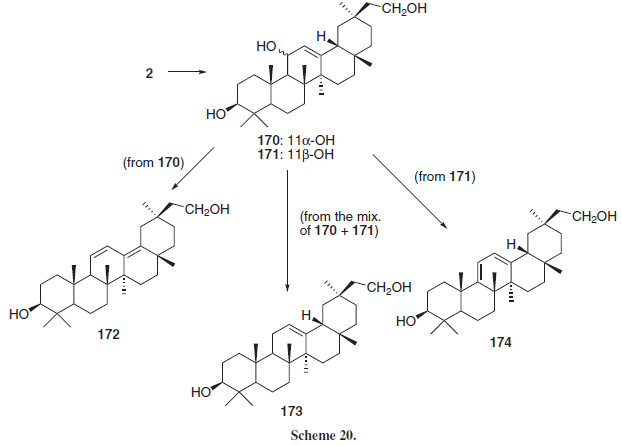
Ablise et al.27 reduced 2 in order to obtain a 11- and 30-diol (Scheme 20, conditions not shown in reference) as a mixture of epimers 11α and 11β (170 and 171, respectively) that were reacted as a mixture in order to generate the 11-deoxo compound 173 or separately, leading to the conjugated dienes 172 and 174, respectively. The synthesized compounds presented in vitro antioxidant activity in a rat microsomal cytochrome P450/NADPH system, being 170 and 171 the most active, inhibiting in 50 and 51% (respectively) the formation of reactive oxygen species, followed by 172 (41%) and 174 (44%) indicating, according with the authors, that chemical reduction of the 11-keto and 30-carboxylic acid groups lead to compounds with greater antioxidant activity.
3.7. Strategies for new GLA derivatives
Some syntheses reported at the literature were performed in order to explore the chemical versatility of GLA and, even without pharmacological activities associated to them yet, may constitute as valuable information to the medicinal chemists as they pinpoint new chemical strategies for obtaining new bioactive GLA derivatives. The following syntheses are classified by the GLA rings being modified.
3.7.1. Modifications at rings A and C
Mikhailova et al.28 performed the ozonolysis reaction of 11-desoxo-GLA (137), obtaining the 3,12-dione derivative 175 using CH2Cl2 as solvent or the 12-one derivative 96 using a mixture of CH2Cl2:MeOH as solvent (Scheme 21).
The same research group employed the 30-methyl ester of 137 (176) as starting material for the synthesis of A-nor-derivatives through the Wagner-Meerwein rearrangement. Depending on the reaction temperature, two different products were obtained (177 and 179), as shown in Scheme 22. Partial ozonolysis of 177 gave the epoxide 178 as product, and oxidation of 179 with RuO4 gave 180 and 181 as a mixture (2:1) of products, while partial ozonolysis of 179 gave only 180 as product.29
3.7.2. Modifications at rings A and E
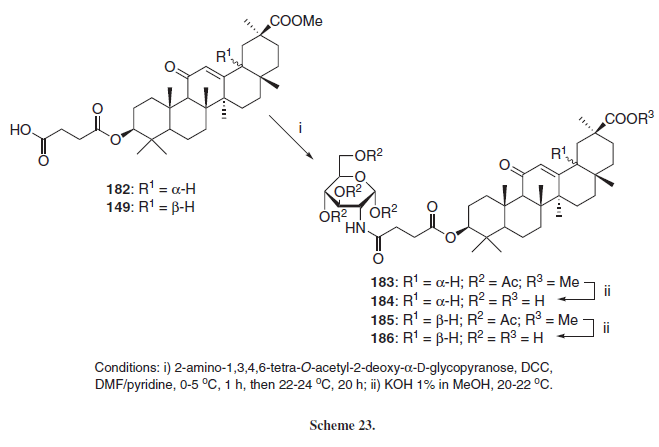
Employing 18-α (182) or 18-β (149) hemisuccinyl esters of GLA, Kondratenko et al.30 synthezised new glycopyranosyl amides employing an aminosugar derivative and DCC as coupling promoter (Scheme 23), leading to the respective amides 183 and 185, which were reacted with KOH in order to generate the un-protected derivatives 184 and 186, respectively.
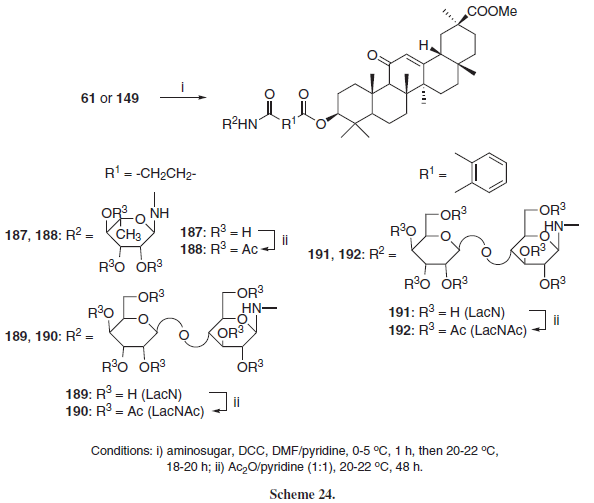
Mustafina et al.31 further expanded this family of compounds employing the hemiphtalyl and hemisuccinyl esters of GLA (61 and 149, respectively) leading to the N-glycoconjugates 187-192 (Scheme 24).
3.7.3. Modifications at rings C and E
Yong et al.32 employed 2 or its 11-deoxy derivative 137 (obtained through the Clemmensen reduction of 2) to synthesize a series of 4 derivatives (193-196, Scheme 25).
3.7.4. Modifications at ring E
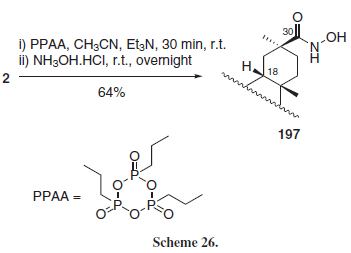
Ech-Chalad et al.33 employed the cyclic phosphonic anhydride PPAA as a carboxylic acid activating agent for hydroxamation reactions in a series of carboxylic acids, including GLA (2) (Scheme 26), obtaining selectively the 30-hydroxamic acid 197.
Two sequential couplings involving GLA (2), 1,6-hexanediamine and biotin (Scheme 27) generated the GLA-hexenediamine-biotin conjugate 198, reported by Shi et al.34
Liu et al.35 synthesized novel GLA-isoxazole derivatives 203 and 204 via DCC-promoted coupling of the isoxazole amines 201 and 202, respectively (Scheme 28).
4. New Pharmacological Activities Reported for GL and GLA
In the period covered by this review, some new pharmacological activities were reported for GL (1), such as: antiviral (against flavivirus36, HIV37 and SARS-coronavirus38) antiallergic,39 antithrombotic in experimental models in rats,40,41 capable of lowering hepatocellular carcinogenesis rates in patients with interferon-resistant active chronic hepatitis C,42 presenting a protective in vitro effect in gastric epithelial cells against H2O2-induced cell death43 and as an inhibitor of influenza A virus uptake into human lung cells.44 GL and GLA (2) were also reported as possessing a protective effect on acute cholestasis induced by α-naphtyl isocyanate in rats45 and GLA was reported as an apoptosis inductor in human hepatoma, promyelotic leukemia and stomach cancer cells,46 as an antihyperglycemic agent in rats with streptozocin-induced diabetes,47 and as presenting antiviral activity against herpes simpex virus 1 (HSV-1).48
Although several interesting biological activities are known for these two compounds, few molecular targets are described for them. GL (1) is described as an inhibitor of the high-mobility group box 1 protein (HMGB1), a signaling molecule involved in acute and chronic inflammation. GL inhibits the chemoattractant and mitogenic activities of HMGB1, but not its intranuclear DNA-binding function.49 GL is also described as an inhibitor of thrombin, a protease involved in the blood clotting cascade.40 As mentioned before, the anti-inflammatory activity of GL and GLA is accounted for the inhibition of the β-hydroxysteroid dehydrogenase enzyme 2 (β-HSD-2), an enzyme responsible for the conversion of the glucocorticoid hormone cortisol in its active form cortisone.3, 4
5. Concluding Remarks
Several different synthetic strategies and some pharmacological activities were highlighted in this review, employing GL or its aglycon, GLA, mainly from research groups in Russia, Korea and Japan. As for the synthetic strategies, most of the reactions described consisted of functionalizations and/or chemical derivatizations of the functional groups already present in both compounds, and can be summarized as: (i) amide or ester synthesis involving the carboxyl groups at C-30 (GL or GLA) or both of the glucoronic acids (GLA); (ii) ester synthesis, oxidation or elimination of the hydroxyl group at C-3; (iii) further functionalization of the 3-keto derivative; (iv) reduction of the ketone group at C-11 and/or reduction of the carboxyl group at C-30; and (v) insaturation of rings A and/or C and further modification of the same. Such chemical strategies are straightforward and easily applicable to these compounds and other related triterpenes. Also, in some of the described reactions it is possible to observe some chemoselectiviy (ex.: Schemes 2 and 3) between similar functional groups in the compounds.
It is noteworthy that GL and GLA, as well as its derivatives described in this review, presented a plethora of pharmacological activities, such as: antiviral, immunomodulating (inducing or suppressing immune response), antiallergic, anti-ulcer, antibacterial (against H. pylori), as PPAR-γ agonists, anti-inflammatory, human 20S proteasome-ubitquitin complex inhibitors, cytotoxic and antiproliferative, antioxidant and 11β-HSD-1 and 2 inhibitors, some with selectivity for one of the subtypes of the enzyme. Such broad range of activities is, at first, undesirable in the choice or identification of a new lead compound. However, the pharmacological activities presented by the discussed derivatives may be associated to specific pharmacophoric groups, such as the cytotoxic activity being related to the presence of a double bond between C-1 and C-2 and a cyano group in C-2 and the presence of glycosyl substituents conjugated with aminoacids and/or other carbohydrates at C-3 leading to derivatives with anti-viral activity. With additional studies, new pharmacophoric groups may unravel guides to selectively explore GL and GLA as scaffolds for diverse sets of pharmacological activities. Alternatively, as multifunctional compounds can be conceived as molecules with improved efficacy,50 some activities described above can be conjugated in a synergic way in order to achieve a better pharmacological profile, such as the antiviral and the immunostimulating activities of some GLA-amino acid conjugates, shown in Schemes 1 through 4. Ultimately, GL and GLA may be considered for its several pharmacological activities as privileged structures,51 and potentially useful as scaffold for the design of new pharmacologically active compounds.

Cedric Stephan Graebin has recieved his PhD degree from the Federal University of Rio Grande do Sul (UFRGS - 2008) and completed his postdoctoral training with Prof. Jorge A. Guimarães (UFRGS - 2009). He is an Adjunct Professor at the Departament of Chemistry, Federal University Rural of Rio de Janeiro. His research interest is the synthesis of new pharmacologically active compounds from both solution and solid-phase synthesis.

Hugo Verli has received his PhD degree from the Federal University of Rio Grande do Sul (UFRGS - 2005) and completed his postdoctoral training with Prof. Jorge A. Guimarães (2009). He is an Adjunct Professor at the Faculty of Pharmacy (UFRGS). His research interests include Medicinal Chemistry and Molecular Modelling, with enphasis in macromolecules structure, conformation and function.

Jorge A. Guimarães has received his PhD degree from the Federal University of São Paulo (Unifesp - 1972) and completed his postdoctoral training at the National Institutes of Health (USA - 1975). He is currently a Full Professor at the Biotechnology Center, Federal University of Rio Grande do Sul, working in Protein Chemistry and Enzymology areas, with emphasis on the isolation and characterization of novel biologically active natural products, proteolytic enzymes and toxic proteins, as well as their relationships to the haemostatic process. Until now, Professor Guimarães has published around 140 full papers in indexed journals.
Submitted: February 19, 2010
Published online: June 18, 2010
- 1. Kitagawa, I.; Pure Appl. Chem. 2002, 74, 1189.
- 2. Fiore, C.; Eisenhut, M.; Ragazzi, E.; Zanchin, G.; Armanini, D. ; J. Ethnopharmacol. 2005, 99, 317.
- 3. Baltina, L. A.; Curr. Med. Chem. 2003, 10, 155.
- 4. Williams, G.; Leclerq, C.; Knaap, A. G. A. C.; WHO Food Additives Series 54: Safety and Evaluation of Certain Food Additives; WHO: Geneva, 2006.
- 5. Baltina, L. A.; Rhizova, S. A.; Vasil'eva, E. V.; Tolstikov, G. A.; Sakhaudtinova, G. M.; Zarudii, F. S.; Russ. J. Bioorg. Chem.. 1996, 22, 926.
- 6. Kondratenko, R. M.; Baltina Jr., L. A.; Baltina, L. A.; Baschenko, N. Zh.; Tolstikov, G. A.; Russ. J. Org. Chem. 2006, 32, 595.
- 7. Kondratenko, R. M.; Baltina, L. A.; Vasil'eva, E. V.; Baltina Jr., L. A.; Ismagilova, A. F.; Nasyrov, Kh. M.; Baschenko, N. Zh.; Kireeva, R. M.; Fridman, S. M.; Tolstikov, G. A.; Russ. J. Bioorg. Chem. 2004, 30, 53.
- 8. Kondratenko, R. M.; Baltina, L. A.; Vasil'eva, E. V.; Nasyrov, Kh. M.; Kireeva, R. M.; Baschenko, N. Zh.; Fridman, S. M.; Baltina Jr., L. A.; Tolstikov, G. A.; Russ. J. Org. Chem. 2004, 30, 148.
- 9. Baltina Jr., L. A.; Kondratenko, R. M.; Baltina, L. A.; Plyasunova, O. A.; Galin, F. Z.; Tolstikov, G. A.; Chem. Nat. Compd. 2006, 42, 543.
- 10. Kondratenko, R. M. Baltina, L. A.; Mustafina, S. R.; Vasil'eva, E. V.; Pompei, R.; Deidda, D.; Plyasunova, O. A.; Pokrovskii, A. G.; Tolstikov, G.; Russ. J. Bioorg. Chem. 2004, 30, 275.
- 11. Hoever, G.; Baltina, L.; Michaelis, M.; Kondratenko, R.; Baltina, L.; Tolstikov, G. A.; Doerr, H. W.; Cinatl, J.; J. Med. Chem 2005, 48, 1256.
- 12. Park, H-Y.; Park, S-H.; Yoon, H.; Han, M. J.; Kim, D-H.; Arch. Pharmacal Res. 2004, 27, 57.
- 13. Kondratenko, R. M. ; R. M. Baltina, L. A.; Mustafina, S. R.; Vasil'eva, E. V.; Ismagilova, A. F.; Vasil'eva, N. G.; Tolstikov, G. A.; Russ. J. Bioorg. Chem. 2003, 6, 601.
- 14. Krausse, R.; Bielenberg, J.; Blaschek, W.; Ullmann, U.; J. Antimicrob. Ther. 2004, 54, 243.
- 15. Yu, D.; Sakurai, Y.; Chen, C.; Chang, F-R.; Kashiwada, Y.; Lee, K-H.; J. Med. Chem. 2006, 49, 5462.
- 16. Chintharlapalli, S.; Papineni, S.; Jutooru, I.; McAlees, A.; Safe, S.; Mol. Cancer Ther. 2007, 6, 1588.
- 17. Liu, D.; Song, D.; Guo, G.; Wang, R.; Lv, J.; Jing, Y.; Zhao, L.; Bioorg. Med. Chem. 2007, 15, 5432.
- 18. Honda, T.; Round, B. V.; Bore, L.; Finlay, H. J.; Favaloro, F.G.; Suh, N.; Wang, Y.; Sporn, M. B.; Gribble, G. W.; J. Med. Chem. 2000, 43, 4233.
- 19. Chadalapaka, G.; Jutooru, I.; McAless, A.; Stefanac, T.; Safe, S.; Bioorg. Med. Chem. Lett. 2008, 18, 2633.
- 20. Nakagawa-Goto, K.; Nakamura, S.; Bastow, K. F.; Nyarko, A.; Peng, C-Y.; Lee, F-Y.; Lee, F-C.; Lee, K-H.; Bioorg. Med. Chem. Lett. 2007, 17, 2894.
- 21. Tatuzaki, J.; Taniguchi, M.; Bastow, K. F.; Nakagawa-Goto, K.; Morris-Natschke, S.; Itokawa, H.; Baba, K.; Lee, K-H.; Bioorg. Med. Chem. 2007, 15, 6193.
- 22. Maitraie, D.; Hung, C.; Tu, H.; Liou, Y.; Wei, B.; Yang, S.; Wang, J.; Lin, C.; Bioorg. Med. Chem. 2009, 17, 2785.
- 23. Su, X.; Vicker, N.; Lawrence, H.; Smith, A.; Purohit, A.; Reed, M. J.; Potter, B. V. L.; J. Steroid Biochem. Mol. Biol. 2007, 104, 312.
- 24. Vicker, N.; Su, X.; Lawrence, H.; Cruttenden, A.; Purohit, A.; Reed, M. J.; Potter, B. V. L.; Bioorg. Med. Chem. Lett. 2004, 14, 3263.
- 25. Huang, L.; Yu, D.; Ho, P.; Qian, K.; Lee, K-H.; Chen, C.; Bioorg. Med. Chem. 2008, 16, 6696.
- 26. Bang, E-K.; Kim, B. H.; Tetrahedron Lett. 2009, 50, 2545.
- 27. Ablise, M.; Leininger-Muller, B.; Wong, C. D.; Siest, G.; Loppinet, V.; Visvikis, S.; Chem. Pharm. Bull 2004, 52, 1436.
- 28. Mikhailova, L. R.; Khudobko, M. V.; Baltina, L. A.; Kukovinets, O. S.; Mavrodiev, V. K.; Galin, F. Z.; Chem. Nat. Compd. 2007, 43, 571.
- 29. Mikhailova, L. R.; Baltina, L. A.; Kondratenko, R. M.; Kunert, O.; Spirikhin, L. V.; Galin, F. Z.; Tolstikov, G. A.; Chem. Nat. Compd. 2006, 42, 553.
- 30. Kondratenko, R. M.; Mustafina, S. R.; Baltina, L. A.; Galin, F. Z.; Tolstikov, G. A.; Chem. Nat. Compd. 2005, 41, 7.
- 31. Mustafina, S. R. ; Baltina Jr., L. A.; Kondratenko, R. M.; Baltina, L. A.; Galin, F. Z.; Tolstikov, G. A.; Chem. Nat. Compd. 2006, 42, 67.
- 32. Yong, J-P.; Wang, J. W.; Aisa, H. A.; Liu, L. J.; Chem. Nat. Compd. 2008, 44, 194.
- 33. Ech-Chalad, A.; Minassi, A.; Berton, L.; Appendino, G.; Tetrahedron Lett. 2005, 46, 5113.
- 34. Shi, J.; Zhou, Y.; He, M.; Jin, J.; Xiao, Y.; Xiao, J.; Wei, D.; Chem. Nat. Compd. 2006, 42, 325.
- 35. Liu, L; Yong, P.; Dai, X; Jia, J.; Wang, X; Wang, J.; Chem. Res. Chin. Univ. 2006, 27, 1669.
- 36. Crance, J. M.; Scaramozzino, N.; Jouan, A.; Garin, D.; Antiviral Res. 2003, 58, 73.
- 37. Harada, S.; Biochem. J. 2005, 392, 191.
- 38. Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H. W.; Lancet 2003, 361, 2045.
- 39. Ram, A; Mabalirajan, U.; Das, M.; Bhattacharya, I.; Dinda, A. K.; Gangal, S. V.; Ghosh, B.; Int. Immunopharmacol. 2006, 6, 1468.
- 40. Mendes-Silva, W.; Assafim, M.; Ruta, B.; Monteiro, R. Q.; Guimarães, J. A.; Zingali, R. B.; Thromb. Res. 2003, 112, 93.
- 41. Nakata, N; Kira, Y; Yabunaka, Y; Takaoka, K.; J. Orthop. Sci. 2008, 13, 456.
- 42. Ikeda, K.; Arase, Y.; Kobayashi, M.; Saitoh, S.; Someya, T.; Hosaka, T.; Sekazi, H.; Akuta, N.; Suzuki, Y.; Suzuki, F.; Kumada, H.; Dig. Dis. Sci. 2006, 51, 603.
- 43. Oh, H-M.; Lee, S.; Park, Y-N.; Choi, E.; Choi, J.; Kim, J.; Kweon, J.; Han, W-C.; Choi, S.; Han, J-K.; Son, J-K.; Lee, S-H.; Jun, C.; Exp. Biol. Med. 2009, 234, 263.
- 44. Wolkerstorfer, A; Kurz, H; Bachhofner, N; Szolar, O. H. J.; Antiviral Res. 2009, 83, 171.
- 45. Zhai, D.; Zhao, Y.; Chen, X.; Guo, J.; He, H.; Yu, Q.; Yang, J.; Davey, A. K.; Wang, J.; Planta Med. 2007, 73, 1.
- 46. Hibasami, H.; Iwase, H.; Yoshioka, K.; Takahashi, H.; Int. J. Mol. Med. 2006, 17, 215.
- 47. Kalaiarasi, P.; Pugalendi, K. V.; Eur. J. Pharmacol. 2009, 606, 269.
- 48. Ikeda, T.; Yokomizo, K.; Okawa, M.; Tsuchihashi, R.; Kinjo, J.; Nohara, T.; Uyeda, M.; Biol. Pharm. Bull. 2005, 28, 1779.
- 49. Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M. E.; Chem. Biol. 2007, 14, 431.
- 50. Cavalli, A.; Bolognesi,M. L.; J. Med. Chem. 2009, 52, 7339.
- 51. Duarte, C. D.; Barreiro, E. J.; Fraga, C. A. M.; Mini-Rev. Med. Chem. 2007, 7, 1108.
Publication Dates
-
Publication in this collection
10 Sept 2010 -
Date of issue
2010
History
-
Received
19 Feb 2010 -
Accepted
18 June 2010