Abstract
Concentrations and identity of ions in the soil solution may affect soil phosphorus (P) reactions and P availability. In this study, the magnitude of these reactions was evaluated following the application of Minjingu phosphate rock (MPR) combined with chloride and carbonate salts of Na and Ca within an incubation experiment. Twenty-one days later NaOH-P and HCl-P were determined. This investigation was undertaken with the aim of identifying the role of Ca-ion activity in the liquid phase on the solubilization of MPR and formation of insoluble Ca-P phases. The increase in pH was higher with Na2CO3 than with CaCO3, while both CaCl2 and NaCl resulted in slight decreases in pH. The dissolution of MPR was higher overall when MPR was applied singularly than for the combined application of the phosphate rock with salts of calcium or sodium after 60 days of incubation. Dissolution of MPR decreased as levels of CaCO3 or CaCl2 increased but the decrease was more pronounced in CaCO3-treated than in CaCl2-treated soils. Ca-ion activity in the liquid phase is the main factor responsible for the insolubilization of MPR and the formation of insoluble Ca-P phases (HCl P). The formation of Ca-P solid phases increased with the concentration of Ca-ions, and was governed by the pH and nature of the accompanying anion. For soils with low levels of exchangeable cations and where liming is a recommended intervention measure, Ca from lime will form insoluble P phases and reduce the dissolution of PR and P availability to plants.
Phosphorus; anion; cation; calcium
Introduction
In Sub-Saharan Africa (SSA), phosphorus is identified as the major limiting nutrient in the vast majority of soils (Bationo et al., 1997Bationo A., E. Ayuk, D. Ballo, and M. Koné. 1997. “Agronomic and Economic Evaluation of Telemsi Phosphate Rock in Different Agroecological Zones of Mali.” Nutrient Cycling in Agroecosystems, 48:179-189.). Such soils constitute up to 55 % of the agricultural land in SSA (Bationo et al., 1986Bationo, A.; Mughogho, S.K.; Mokwunye, A.U. 1986. Agronomic evaluation of phosphate fertilizers in tropical Africa. p. 283-318. In: Mokwunye, A.U.; Vlek, P.L.G., eds. Management of nitrogen and phosphorus fertilizers in sub-Saharan Africa. Martinus Nijhoff, Dordrecht, The Netherlands.). In Kenya, for example, Braun et al. (1997)Braun, A.R.; Smaling, E.M.A.; Muchugu, E.I.; Shepherd, K.D.; Corbett, J.D., eds. 1997. Maintenance and Improvement of Soil Productivity in the Highlands of Ethiopia, Kenya, Madagascar and Uganda: An Inventory of Spatial and Non-Spatial Survey and Research Data on Natural Resources and Land Productivity; African Highlands Initiative. International Centre for Research in Agroforestry, Nairobi, Kenya. (AHI Technical Report Series, 6). noted that in addition to P deficiency, applied P was susceptible to fixation in about 66 % of the soils in the highlands. Low content of available P in these soils is genetic and agriculture only tends to aggravate the process. The main strategy to cope with P deficiency in the tropics has been the addition of fertilizers, either in the form of synthetic fertilizer or in the form of phosphate rock (PR) (Walker and Syers, 1976Walker, T.W.; Syers, J.K. 1976. The fate of phosphorus during pedogenesis. Geoderma 15: 1–19.; Zapata and Zaharah, 2002Zapata, F.; Zaharah, A.R. 2002. Phosphorus availability from phosphate rock and sewage sludge as influenced by the addition of water-soluble phosphate fertilizer. Nutrient Cycling in Agroecosystems 63: 43-48.).
When any phosphate fertilizer is applied to the soil, several processes are initiated to control the P dynamics. The extent of these reactions is influenced by the physical and chemical properties of the soil (Sample et al., 1980Sample, E.C.; Soper, R. J.; Racz, J.G. 1980. Reactions of phosphate fertilizers in soils. p. 263-310. In: Khasawneh, F.E.; Sample, E.C.; Kamprath, E.J., eds. The role of phosphorus in agriculture. ASA/CSSA/SSSA, Madison, WI, USA.). Water soluble fertilizers such as superphosphates are expensive and many of the less-developed countries are exploring the possibility of making maximum use of indigenous agro-minerals such as PRs, as substitutes for expensive, imported fertilizers. Therefore, an accurate determination of the PR dissolution in soils is of major importance.
Dissolution of PR in soil solution requires an adequate supply of moisture and protons (H+), as well as the removal of reactive products such as Ca2+, H2PO4−, and F− from the site of reaction. The rate of dissolution is considered to depend on the lowering of Ca2+ and H2PO4− activities (Bolan et al., 1990Bolan, N.S.; White, R.E.; Hedley, M.J. 1990. A review of the use of phosphate rocks as fertilizers for direct application in Australia and New Zealand. Australian Journal of Experimental Agriculture 30: 297-313.; Chien et al., 1980a, b; Kanabo and Gilkes, 1987Kanabo, I.; Gilkes, R.J. 1987. The role of soil pH in the dissolution of phosphate rock fertilizers. Fertilizer Research 12: 165-174.; Khasawneh and Doll, 1978Khasawneh, F.E.; Doll, E.C. 1978. The use of phosphate rock for direct application to soils. Advances in Agronomy 30: 159–206.; Robison et al., 1992a, b; Robinson and Syers, 1990Robinson, J.S.; Syers, J.K. 1990. A critical evaluation of factors influencing the dissolution of Gafsa phosphate rock. Journal of Soil Science 41: 597-605.; Wilson and Ellis, 1984Wilson, M.A.; Ellis, B.G. 1984. Influence of calcium solution activity and surface area on the solubility of selected rock phosphates. Soil Science 138: 354–359.). Several researchers have studied the availability of P from PR sources applied to soil (Bolan et al., 1990Bolan, N.S.; White, R.E.; Hedley, M.J. 1990. A review of the use of phosphate rocks as fertilizers for direct application in Australia and New Zealand. Australian Journal of Experimental Agriculture 30: 297-313.; Bolland and Gilkes, 1997Bolland, M.D.A.; Gilkes, R.J. 1997. The agronomic effectiveness of reactive phosphate rocks, 2. Effect of phosphate rock reactivity. Australian Journal of Experimental Agriculture 37: 937-946.; Chien et al., 1987Chien, S.H.; Adams, F.; Khasawneh, F.E.; Henao, O. 1987. Effects of combinations of triple superphosphate and a reactive phosphate rock on yield and phosphorus uptake by corn. Soil Science Society of America Journal 51: 1656-1658.; Engelstad et al., 1974Engelstad, O.P.; Jugsujinda, A.; De Datta, S.K. 1974. Response by flooded rice to phosphate rocks varying in citrate solubility. Soil Science Society of America Proceedings 38: 524-529.); however, studies on the interaction effect of PR and different ion combinations and how they affect P availability and extractability are limited.
Considerable attention has been given towards the effect of liming on the agronomic effectiveness of PR materials in soil (He et al., 1996aHe, Z.L.; Baligar, V.C.; Martens, D.C.; Ritchey, K.D.; Kemper, W.D. 1996a. Kinetics of phosphate rock dissolution in an acid soil amended with liming materials and cellulose. Soil Science Society of America Journal 60: 1589-1595. and bHe, Z.L.; Baligar, V.C.; Martens, D.C.; Ritchey, K.D.; Kemper, W.D. 1996b. Factors affecting phosphate rock dissolution in an acid soil amended with liming materials and cellulose. Soil Science Society of America Journal 60: 1596-1601.; Baligar et al., 1997Baligar, V.C.; He, Z.L.; Martens, D.C.; Ritchey, K.D. 1997. Effect of phosphate rock, lime, and cellulose on ryegrass in an acidic soil. Plant and Soil 195: 129-136.). From a practical standpoint it is interesting to consider how P interacts as a PR compared to soluble P fertilizer in limed acid soils. In this study, the interaction effect of combinations of Minjingu PR (MPR) and calcium and sodium as carbonate or chloride salt on P release was investigated. This can help clarify the relative importance of carbonate solid phases and other soil minerals in inducing PR partitioning between the dissolved and the solid phases. The study also investigated how the slurry concentration, pH and electrolyte concentration influence P-extraction by sequential and resin methods.
Materials and Methods
Superficial samples from a Kandiudalfic Eutrudox from Maseno in western Kenya were used. A number of the properties of the soil are presented in Table 1. Phosphorus sorption isotherms determined by theFox and Kamprath (1970)Fox, R.L.; Kamprath E.J. 1970. Phosphate sorption isotherms for evaluating the phosphate requirements of soil. Soil Science Society of America Proceedings 34: 902-907. method indicated that a soil solution P concentration of 0.2 mg P L−1 corresponded to 260 mg sorbed P kg−1. The soil samples were air dried, ground and passed through a 2 -mm sieve.
Minjingu phosphate rock (MPR) (12.8 % total P (128 g kg−1), 3.0 % neutral ammonium citrate soluble P (30 g kg−1), and 26.9 % Ca (269 g kg−1) from northern Tanzania, and triple superphosphate (TSP) (20 % P) (200 g kg−1) were the sources of P used in this study. MPR was applied as finely ground powder (> 90 % passing through a 100-mesh sieve), and TSP was applied in commercially available form after hand grinding to a fine powder.
Incubation
Bulk dry soil samples were separately incubated with 5 levels of analytical grade CaCO3, CaCl2, Na2CO3 and NaCl at 0 (deionized water), 20, 40, 50 or 80 mmolc kg−1 (0, 1.6, 3.3, 4.1 or 6.6 t ha−1) for 21 and 60 days at 23 ± 2 °C in order to investigate the effects of added cations and anions, separately, on the dissolution of MPR. The soil samples (150 g) were mixed thoroughly with either deionized water (water) or CaCO3, CaCl2Na2CO3 or NaCl in separate plastic bags, moistened to 80 % of field capacity before the incubation. After a period of 21 days the soils were air-dried and ground to pass through a 2 mm sieve.
The P fertilizers were added to 150 g air-dry pre-incubated soil at rates equivalent to 0 and 91 mg P/kg and once thoroughly mixed moistened with deionized water to approximately 80 % of field capacity. Soil samples were incubated at this moisture level in glass bottles at 23 ± 2 °C for 21 days before analysis of resin P (AER and ACER-P) and sequential-P as described below.
Extraction of P
The extent of PR dissolution in soil was determined from increases in inorganic P (Pi) values, or from the P remaining in the undissolved PR. The Pi values were calculated as the difference between the Pi extracted from unamended soil and soil amended with PR. P was extracted by using both sequential and resin P methods. The resin extraction method examined both the anion alone and anion + cation resin systems extractions (Sibbesen, 1978Sibbesen, E. 1978. An investigation of the anion-exchange resin method for soil phosphate extraction. Plant and Soil 50: 305–321.).
Resin extractable P was determined using the following resin systems: (i) anion-exchange resin (Dowex 1-X8) in the bicarbonate form, designated AER; (ii) anion-exchange resin (Dowex 1-X8) (bicarbonate form) plus cation-exchange resin (hydrogen form), designated AER+CER. The cation- and anion-exchange resins were enclosed in separate nylon mesh bags 1 g (moist weight) of resin per bag. Anion resin extractable P (AER-P) was determined by shaking 2.5 g of soil for 16 h with 40 mL of deionized water and one mesh bag containing 1 g (moist weight) anion exchange resin in bicarbonate form (Sibbesen, 1978Sibbesen, E. 1978. An investigation of the anion-exchange resin method for soil phosphate extraction. Plant and Soil 50: 305–321.). Anion plus cation resin extractable P (ACER-P) was determined as for AER-P, with the addition of one mesh bag containing 1 g (moist weight) cation exchange resin in sodium form. The P adsorbed by the AER under both methods was extracted by shaking the resin bag with 20 mL of 0.5 M HCl for 1 h. Phosphorus in the extracts was determined by the molybdenum blue method ofMurphy and Riley (1962)Murphy, J.; Riley, J.P. 1962. A modified single solution method for the determination of phosphorus in natural waters. Analytica Chimica Acta 27: 31-36..
P fractionation
Three extractants were examined for sequential method: (i) 0.5 M sodium hydroxide (0.5 M NaOH) to extract dissolved and recently adsorbed Pi (Mackay et al., 1986MacKay, A.D.; Syers, J.K.; Tillman, R.W.; Gregg, P.E.H. 1986. A simple model to describe the dissolution of phosphate rock in soils. Soil Science Society of America Journal 50: 291-296.); (ii) 0.5 M sodium bicarbonate at pH 8.5 (0.5 M NaHCO3) to extract dissolved Pi (Olsen et al., 1954Olsen, S.R.; Cole, C.V.; Watanabe, F.S.; Dean, L.A. 1954. Estimation of vailable phosphorus in soils by extraction with sodium bicarbonate. USDA, Washingtom, DC, USA. (Circular, 939).); and (iii) 1 M hydrochloric acid (1 M HCl) to extract precipitated Ca-P and undissolved PR (Apthorp et al., 1987Apthorp, J.N.; Hedley, M.J.; Tilman, R.W. 1987. The effect of nitrogen fertilizer form on the plant availability of phosphate from soil, phosphate rock and monocalcium phosphate. Fertilizer Research 12: 269-284.).
Sequentially extractable P with and without NaCl prewash was determined for treatments from 21 and 30 days by a modification of the method of Tiessen and Moir (1993)Tiessen, H.; Moir, J.O. 1993. Characterization of available phosphorus by sequential extraction. p. 76–86. In: Carter, M.R., ed. Soil sampling and methods of analysis. Lewis, Boca Raton, FL, USA.. The modification involved omitting the initial resin extraction step due to the substantial solubility of solid MPR in the resin-water-soil system (Mutuo et al., 1999Mutuo, P.K.; Smithson, P.C.; Buresh, R.J.; Okalebo, R.J. 1999. Comparison of phosphate rock and triple superphosphate on a phosphorus-deficient Kenyan soil. Communication in Soil Science & Plant Analysis 30: 1091–1103.). Briefly, 0.5 g of soil was shaken for 16 h with 30 mL of 0.5 M NaHCO3 (pH 8.5). Samples were centrifuged at 5,700 x g for 10 min; an aliquot of the supernatant was acidified with 0.6 mL of 1.2 M H2SO4 and centrifuged for 10 min to precipitate organic matter.
The supernatant was then analyzed for inorganic P (Pi). A second aliquot was digested with acidified ammonium per sulfate in an autoclave (103 k Pa, 121 °C) for 1 h and then analyzed for total P (Pt). Extractable organic P (Po) was calculated as the difference between Pt and Pi. The soil residue was extracted with 30 mL of 0.1 M NaOH for 16 h. Aliquots were analysed for Pi and Pt as for the bicarbonate extract. The soil residue was then extracted with 30 mL of 1 M HCl for 16 h, centrifuged as above and an aliquot analysed for Pi. P was analysed by the molybdenum blue method of Murphy and Riley (1962)Murphy, J.; Riley, J.P. 1962. A modified single solution method for the determination of phosphorus in natural waters. Analytica Chimica Acta 27: 31-36..
For the measurement of changes in NaOH and HCl Pi fractions following treatment applications, the soil was prewashed with 1 M NaCl for half an hour at a solid: solution ratio of 1:100. Pre-washing with 1 M NaCl has been proposed by Mackay et al. (1986)MacKay, A.D.; Syers, J.K.; Tillman, R.W.; Gregg, P.E.H. 1986. A simple model to describe the dissolution of phosphate rock in soils. Soil Science Society of America Journal 50: 291-296. to remove both the solution and exchangeable Ca which otherwise may form Ca(OH)2 and CaCO3 during extraction with 0.5 M NaOH and 0.5 M NaHCO3, respectively, and readsorb, or coprecipitate with some of the dissolved P. After centrifuging at 10,000 rpm for 10 min, the supernatant solution was filtered using a 0.45 µm filter and analysed for Pi (Murphy and Riley, 1962Murphy, J.; Riley, J.P. 1962. A modified single solution method for the determination of phosphorus in natural waters. Analytica Chimica Acta 27: 31-36.). The soil residue was subsequently shaken with 0.5 M NaOH for 16 h at a solid:solution ratio of 1:100. After centrifuging and filtering, the solution was analysed for Pi. Phosphorus in the extracts was determined by the molybdenum blue method of Murphy and Riley (1962)Murphy, J.; Riley, J.P. 1962. A modified single solution method for the determination of phosphorus in natural waters. Analytica Chimica Acta 27: 31-36..
Statistical Analysis
Statistical analyses were conducted with the general linear procedure of an SAS program (SAS Statistical Analysis System, version 6.1, 1994). An LSD test was conducted to determine significant differences of the mean values between treatments. Reference to statistical significance refers to p< 0.05 unless otherwise noted.
Results and Discussion
Effect of cations and anions on dissolution of Minjingu PR
Irrespective of the P extraction method, the ability of each salt to increase P in soil solution was associated with the identity of the salt, cation and anion concentration (Table 2). The two extraction methods (sequential and resin) did not always produce the same results in percentage dissolution for the same treatments. Total recovery of the P added by both methods of extraction was less than the P added for all the treatments, although most of the P was recovered with sequential extraction.
There was a marked difference in the extent of dissolution of MPR treated with NaCl, Na2CO3, CaCl2, and CaCO3. For sequential extraction, P recovery followed the order: MPR only > MPR+Na2CO3 > MPR+NaCl > MPR+CaCl2> MPR+CaCO3, while resin P followed the order: MPR+Na2CO3 > MPR only > MPR+CaCO3> MPR+NaCl > MPR+CaCl2. The influence of NaCl, Na2CO3, CaCl2, and CaCO3 on the rate and extent of MPR dissolution can be clearly (Figures 2A and B). In this case MPR was incubated with seen concentrations of NaCl, Na2CO3, CaCl2, and CaCO3 for 21 days. Although the shapes of the P recovery curves were mostly similar for each P fraction, there were large differences in the amounts of MPR recovered in the various Ca and Na treatments. Minimum anion resin P recoveries were observed where chloride salts were added irrespective of the concentration (Figure 1). This may have important implications on the ability of extractants to predict the ability of PR to supply P to plants in soils receiving calcium based amendments e.g. lime.
− Effect of added salt on MPR sequential inorganic P fractions (A) NaOH Pi, (B) HCl Pi (means ± standard error).
Sequential P fractions
The increase in NaOH extractable P was much higher than the increase in Olsen-P (bicarbonate Pi) in the soils amended with MPR (Table 2). This is because the sorbed P and newly formed Fe and Al phosphates can be mostly extracted into 0.5 M NaOH (Hedley et al., 1982Hedley, M.J.; Stewart, W.B.; Chauhan, B.S. 1982. Changes in inorganic and organic soil phosphorus fractions induced by cultivation practices and by laboratory incubations. Soil Science Society of America Journal 46: 970–976.; Tiessen et al., 1984; Wager et al., 1986), but only a fraction can be extracted by the NaHCO3. Consistent with our findings, Alloush (2003)Alloush, G.A. 2003. Dissolution and effectiveness of phosphate rock in acidic soil amended with cattle manure. Plant and Soil 251: 37–46. observed that NaHCO3 extractable P often underestimates PR dissolution. Increased PR dissolution in soils does not guarantee an increase in the amount of plant available P (Sanyal and Datta, 1991Sanyal, S.K.; Datta, S.K.D. 1991. Chemistry of phosphorous transformations in soil. Advances in Soil Science 16: 1–94.). Estimated P from NaHCO3 has a strong correlation with plant P uptake (Zee et al., 1987Zee, S.E.A.T.M. van der; Fokkink, L.G.J.; Riemsdijk, W.H. van. 1987. A new technique for assessment of reversibly adsorbed phosphate. Soil Science Society of America Journal 51: 599–604.;Menon et al., 1989Menon, R.G.; Hammond, L.L,; Sissingh, H.A. 1989. Determination of plant available phosphorus by the iron hydroxide-impregnated filter paper (Pi) soil test. Soil Science Society of America Journal 53: 110–115.; Sharpley, 1991Sharpley, A.N. 1991. Soil phosphorus extracted by iron-aluminum line oxide-impregnated filter paper. Soil Science Society of America Journal 55: 1038–1041.) and is thus usually assumed to be plant available (Hedley et al., 1982Hedley, M.J.; Stewart, W.B.; Chauhan, B.S. 1982. Changes in inorganic and organic soil phosphorus fractions induced by cultivation practices and by laboratory incubations. Soil Science Society of America Journal 46: 970–976.; Tiessen and Moir, 1993Tiessen, H.; Moir, J.O. 1993. Characterization of available phosphorus by sequential extraction. p. 76–86. In: Carter, M.R., ed. Soil sampling and methods of analysis. Lewis, Boca Raton, FL, USA.;Cross and Schlesinger, 1995Cross, A.F.; Schlesinger, W.H. 1995. A literature review and evaluation of the Hedley fractionation: applications to the biogeochemical cycles of soil phosphorus in natural ecosystems. Geoderma 64: 197–214.). On the other hand NaOH-Pi is often assumed to be moderately available (Hedley et al., 1982Hedley, M.J.; Stewart, W.B.; Chauhan, B.S. 1982. Changes in inorganic and organic soil phosphorus fractions induced by cultivation practices and by laboratory incubations. Soil Science Society of America Journal 46: 970–976., Schmidt et al., 1996Schmidt, J.P.; Buol, S.W.; Kamprath, E.J. 1996. Soil phosphorus dynamics during seventeen years of continuous cultivation: Fractionation analysis. Soil Science Society of America Journal 60: 1168–1172.; Ivarsson, 1990Ivarsson, K. 1990. The long-term soil fertility experiments in southern Sweden. IV. Changes in inorganic and organic soil after a pot trial. Acta Agriculturae Scandinavic 40: 205–215.).
Addition of calcium or sodium salts either as carbonate or chloride decreased the concentration of P in the soil solution (NaOH-Pi) but increased that of HCl P (Figure 2A and B). The NaOH Pi recovered decreased as concentrations of salts in carbonate form increased but seemed to reach saturation or stabilize at 40 mmol kg−1 in the case of chloride salts. That is for each sequential P fraction the amount of P recovered was dependent on both the concentration and the nature of accompanying cation and/or anion. More NaOH P was extracted from both sodium salts of carbonate and chloride than from similar salts of calcium irrespective of the concentration (Figure 2A). This seems to agree with the Donnan prediction theory that the activity of the phosphate ion in the vicinity of the adsorbing surface (clay particles, hydroxides of iron and aluminum) should increase as the valency of the saturating cation increases (Wiklander, 1964Wiklander, L. 1964. Cation and anion exchange phenomena. p. 163-205. In: Bear, F.E., ed. Chemistry of the soil. Reinhold, New York, NY, USA.). Thus, the accessibility of the adsorbing surface to phosphorus in solution should be greater with divalent (Ca2+) than with monovalent (Na+) cations in the exchange complex.
Ernani and Barber (1995)Ernani, P.R.; Barber, S.A. 1995. Phosphorus availability in a low pH highly weathered soil as affected by added salts. Ciência Rural 25: 219-222. have also shown that in the presence of different cations the magnitude of P in solution followed the descending order; Na+ > K+ > Mg2+> Ca2+. The increase of NaOH P that occurred after the addition of both sodium carbonate and sodium chloride over both salts of calcium carbonate and chloride was probably due to the diffuse double layer (DDL) effect because Na promotes a thicker DDL relative to calcium, and as DDL becomes thicker it is more difficult for P to approach the adsorption surface.
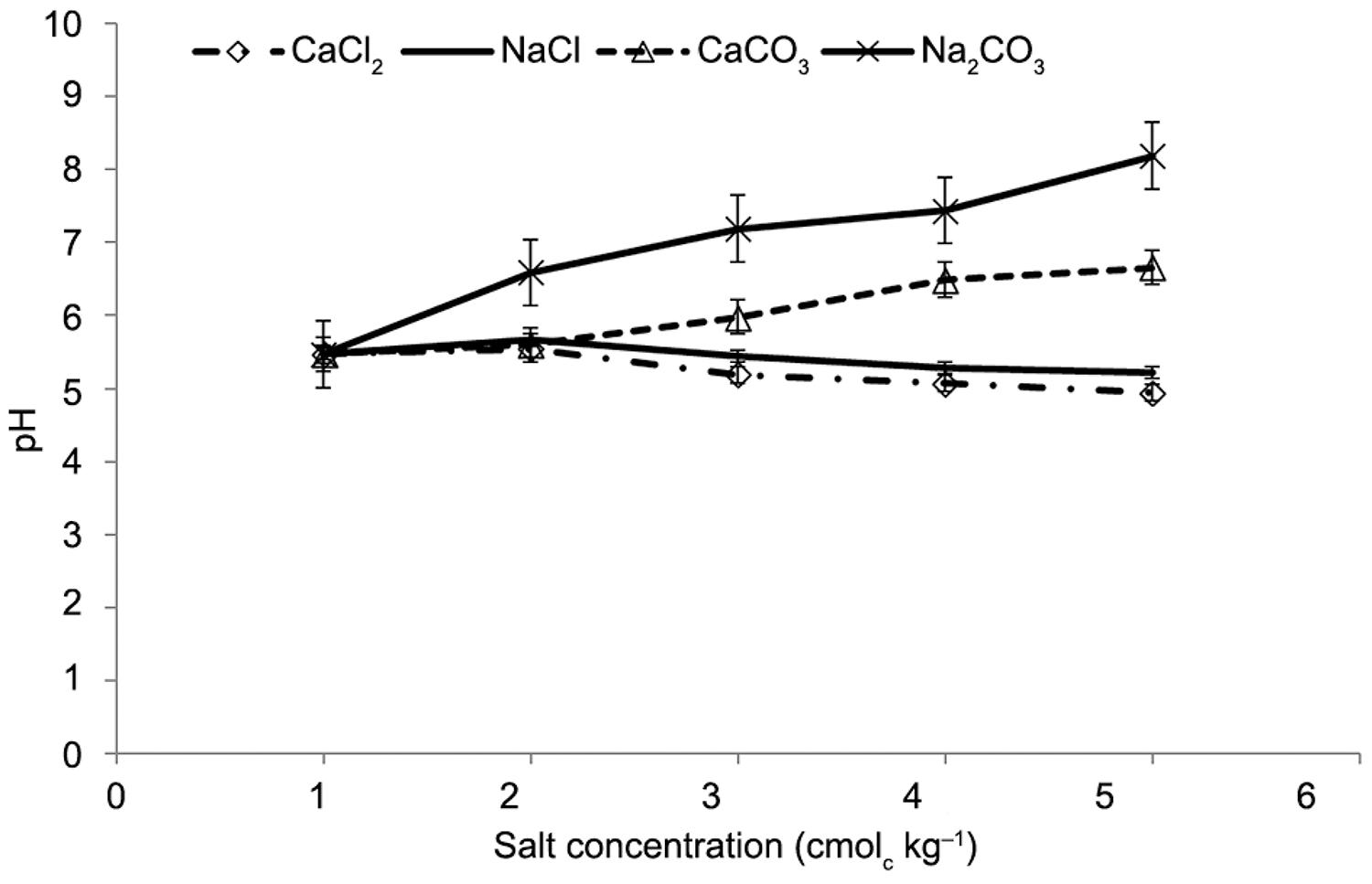
Interestingly more NaOH Pi was extracted from both NaCl and CaCl2treated soils at 80 mmolc kg−1 (Figure 2A). Furthermore, pH values were minimal at this concentration 4.94 and 5.22 for both CaCl2 and NaCl respectively (Figure 3). Bolan and Hedley (1990)Bolan, N.S.; Hedley, M.J. 1990. Dissolution of phosphate rocks in soils. 2. Effect of pH on the dissolution and plant availability of phosphate rock in soil with pH dependent charge. Fertilizer Research 24: 125 134. reported increases in North Carolina phosphate rock, Jordan phosphate rock and Nauru phosphate rock, and dissolution when the soil pH decreased from 6.5 to 3.9. The dissolution of PR can be increased by increasing the supply of protons (H+) or by the continuous removal of the dissolved Ca and P from the dissolution zone (Kirk and Nye, 1986Kirk, G.J.D.; Nye, P.H. 1986. A simple model for predicting the rates of dissolution of sparingly soluble calcium phosphates in soil. III. A predictive model for regularly distributed particles. Journal of Soil Science 37: 511–524.).
At low pH, the most dissolved P is adsorbed by oxides of Al and Fe, or precipitated as Fe and Al phosphates and 0.5 M NaOH extracts most of the P sorbed/precipitated with these components. Because extraction with diluted NaOH removes non occluded Fe-P and Al-P from a soil through desorption or solubilization (Syers et al., 1973Syers, J.K.; Harris, R.F.; Armstrong, D.E. 1973. Phosphate chemistry in lake sediments. Journal of Environmental Quality 2: 1-14.), and because minerals of the apatite group do not dissolve to any significant extent in this reagent (Chang and Jackson, 1957Chang, S.C.; Jackson, M.L. 1957. Fractionation of soil phosphorus. Soil Science 84: 133-144.), any increase in NaOH-extractable P in a soil to which a PR is added should provide an estimate of the amount of P dissolved from the PR. Contrary to NaOH, P, recovery of the HCl fraction increased with increases in salt concentration (Figure 2B). Despite similar shapes of the curves of inorganic P across treatments, there were differences in the magnitude of NaOH and HCl P extracted.
The decrease in NaOH-extractable P in soil to which PR has been added with increasing concentrations of salt indicates decreased dissolution of PR. High base saturation (Ca and Na) also maintains soil P in more stable acid soluble non-extractable forms, keeping contents of the alkaline soluble secondary P forms low. This relationship is due to the stability of Ca-P in highly base saturated soils (Lindsay and Vlek, 1977Lindsay, W.L.; Vlek, P.L.G. 1977. Phosphate minerals. p. 639-672. In: Dixon, J.B.; Weed. S.B., eds. Minerals in soil environments. Soil Science Society of America, Madison, WI, USA.). This is consistent with the increase in the HCl P fraction, which is an estimate of the undissolved PR. The addition of both calcium and sodium salts (carbonates and chlorides) with MPR had significant effects on HCl-Pi concentrations. All rates of CaCl2, CaCO3, NaCl and Na2CO3 application with MPR significantly increased the HCl-Pi concentration in the soil (Figure 2B). This is due to the high concentration of undissolved PR (P associated with Ca) remaining in the soils, which was extracted by HCl.
In general, the magnitude of the increase in HCl-Pi concentrations per unit weight of MPR due to addition of both CaCl2 and CaCO3 was greater than that due to the corresponding Na form of chloride and carbonate. Since dissolution of MPR also releases Ca, according to the mass action law or the common ion effect the dissolution of MPR will be diminished on addition of CaCO3 and CaCl2. The removal of the dissolution reaction products Ca2+, H2PO4− and F− from the site of dissolution and the supply of H+is a driving force for the dissolution of PR, (Khasawneh and Doll 1978Khasawneh, F.E.; Doll, E.C. 1978. The use of phosphate rock for direct application to soils. Advances in Agronomy 30: 159–206.). The HCl-P results in this study are consistent with the observed increase in anion + cation resin extractable P where MPR was applied in combination with either CaCO3 or CaCl2 (Figure 7). The inclusion of cation exchange resin provides a sink for Ca thus decreasing its activity in the soil solution. This increases P concentrations by pulling the dissolution reaction from left to right.
− Effect of carbonate and chloride anions on cation + anion (AER + CER) resin extractable P and anion resin (AER) P (means ± standard error).

pH and MPR solubility
While the carbonate based salts increased the soil pH (alkaline), the pH was depressed (acidic) due to application of chloride salts (Figure 3). The two sources of soil alkalinity (calcium and sodium carbonate) had varying degrees of influence on the solubility of MPR (Figure 4A and B). Where alkalinity was entirely due to calcium, as carbonate, only 27 % of the added MPR dissolved in the soil solution. On the other hand, where soil alkalinity was due to sodium, solubility increased (54 %) over that in the presence of calcium (Figure 4B). In other words, there was a rapid reduction in solubility with increases in pH where alkalinity was due to calcium, and an increase even in excess of the solubility generated by acid reactions, when the pH was raised by the addition of sodium carbonate.
− Relationship between sequential inorganic P fractions and added salt (A) carbonate (B) chloride.
In the presence of calcium carbonate, solubility fell from 46 mg kg–1at pH 5.60 to 13 mg kg–1 at pH 6.66. Solubility in sodium alkalinity was high and it is interesting to note that at pH 7.4 solubility was 31 mg kg–1, in sodium carbonate solution and 13 mg kg–1 for calcium carbonate solution.
As the pH changed, from 5.54 to 4.94 and 5.60 to 6.66 due to the addition of CaCl2 and CaCO3 respectively the dissolution of MPR as NaOH inorganic fraction decreased by 10 % compared to CaCO3 a decrease in CaCO3 of 38 %. Similarly, as the soil pH changed from 5.67 to 5.22 and 6.59 to 8.19 due to additions of NaCl and Na2CO3 respectively, the proportion of the dissolved P extracted by NaOH decreased by 6 % in the NaCl + MPR combination compared to a decline of 37 % when Na2CO3 was applied. This decrease in dissolved P corresponded to an increase in the proportion of HCl inorganic P fraction with an increase in salt concentration. However, the NaOH Pi from MPR plus sodium or calcium chloride relative to that from sodium or calcium carbonate was higher at low pH than at high pH. Furthermore, the amount of NaOH extractable P was more closely related to the nature and amount of added salt cation than to the pH in the soil (Figure 4A and B). It seems that the addition of Na2CO3, reacts with PR (calcium phosphate) to form highly soluble sodium phosphates.
As the pH decreased when both calcium and sodium were added as chloride salts to the soil MPR mixture, there was an increase in NaOH P compared to when carbonate salt of calcium and sodium were added (Figure 4A and B). The decline in pH accompanied by an increase in NaOH extractable P due to the addition of the chloride salts can be attributed to the role of neutral salts in increasing net positive charges that increased the adsorption of applied P (Naidu et al., 1990Naidu, R.; Syers, J.K.; Kirkman, J.H. 1990. Effect of liming on phosphate sorption by acid soils. Journal of Soil Science 41: 165–175.), in the soil. The effect of pH on MPR dissolution was diminished in the presence of sodium carbonate.
For Na-saturated soils, an increase in pH always produced a decrease in phosphate partitioning into the solid phase. This behavior can be explained by increased repulsion between phosphate anions and the surface of variable-charge minerals, such as clay edges and Fe and Al hydroxides, whose negative surface charge increases with increases in pH. For Ca-saturated soils the effect is more complex. The dissolution of PR depends on lowering H2PO4− activity through P sorption which diminishes at higher pH values. The pH affects the adsorption phenomena by influencing the repulsion in the electrical double layer. With increasing Ca concentration in solution, when precipitation becomes predominant, the effect of increasing hydroxyl ions reverses. At higher pH values the solubility of Ca-P mineral phases (MPR) diminishes and precipitation phenomena increase distinctly, resulting in more P being removed from the solution. This is evident in the increase in magnitude of the extractable HCl P (as shown above) and mixed anion and cation resin system (explained below) in the presence of Ca salts.
Effect of added cations and anions on sequential P fractions
With the addition of CO3−2 NaOH P decreased reaching its minimum at 80 mmolc kg−1, while the presence of Cl− gradually decreased NaOH P by up to 40 mmolckg−1 and showed a gradual increase thereafter (Figure 5A). The two anions gradually increased HCl P with as concentration increased although the effect of CO3−2 was stronger than Cl− (Figure 5B). While Cl− anions decreased the soil pH, CO3−2 had the opposite effect and increased the soil pH. The decrease in pH promotes the adsorption of dissolved P by clays, Fe and Al hydroxides due to increases in the positive charge of these soil-colloidal components. The 0.5 M NaOH extracts sorbed P and newly formed Fe and Al phosphates which explain the increase in NaOH-P fraction when Cl− based salts were applied.
− Effect of anion and cation concentration on sequential inorganic P fractions (A) anion (B) cation concentration.
Under conditions such as those in western Kenya, where the soils possess substantial anion exchange capacity (6 to 33 mmolc kg−1) (Hartemink et al., 1996Hartemink, A.E.; Buresh, R.J.; Jama, B.; Janssen, B.H. 1996. Soil nitrate and water dynamics in sesbania fallows, weed fallows and maize. Soil Science Society of America Journal 6: 568−574.), which are more preponderant than those commonly reported for variable charge soils (Hyun et al., 2003; Qafoku and Sumner 2001), more dissolved P will be adsorbed by the anion exchange sites. This is in agreement with the mineralogy of this soil (Table 1) that is dominated by sesquioxides (oxides/hydoxides of Al and Fe) and/or 1:1 clay (kaolinite).
Generally, more NaOH P was extracted where sodium cation was added although it showed a decline as the Na concentration increased (Figure 5B). The increase in NaOH P caused by Na is because Na promotes a thicker double diffuse layer (DDL) relative to Ca, and as DDL becomes thicker it is more difficult for P to approach the adsorption surface.Wiklander (1964)Wiklander, L. 1964. Cation and anion exchange phenomena. p. 163-205. In: Bear, F.E., ed. Chemistry of the soil. Reinhold, New York, NY, USA. found that the thickness of the diffuse double layer is inversely proportional to the valency of the saturating cation. Donnan theory predicts that the activity of the phosphate ion in the vicinity of the clay particles should increase as the valency of the saturating cation increases. Thus, the accessibility of the clay surface to P in an adsorption reaction should be greater with divalent than with monovalent cations in the exchange complex. Pissarides et al. (1968)Pissarides, A.J.; Stewart, W.B.; Reenie, D.A. 1968. Influence of cation saturation on phosphorus adsorption by selected clay minerals. Canadian Journal of Soil Science 48: 151-157. found that phosphate adsorption by three clay minerals (kaolinite, illite, and montmorillonite) saturated with Li, Na, K, Mg, Ca, Sr, and Ba was much higher for divalent than for monovalent cations. The work of Curtin et al. (1987)Curtin, D.; Syers, J. K.; Sraillie, G.W. 1987. The importance of exchangeable cations and resin-sink characteristics in the release of soil phosphorus. Journal of Soil Science 38: 711-716. on 11 acidic (pH 5.5-6.3) New Zealand soils contrasting in P status and retention found that the release of P from Na-saturated soils exceeded that from Ca-dominated soils. They explained this in terms of either enhancing the release of adsorbed phosphate following the removal of exchangeable Ca, increased dissolution of the calcium phosphate phase present as a surface complex, or as a metastable fertilizer reaction product.
Depending on the principles of surface charge balance (Sposito, 2008Sposito, G. 2008. The Surface Chemistry of Soils. Oxford University Press, New York, NY, USA.; Bowden et al., 1980Bowden, J.W.; Posner, A.M.; Quirk, J.P. 1980. Adsorption and charging phenomena in variable charge soils. p. 147-166. In: Theng, B.K.G., ed. Soils with variable charge. New Zealand Society of Soil Science, Lower Hutt, New Zealand.), the electrostatic potential of soil surfaces with a net negative charge will be more negative when the balancing cation is further from the surface. The more the negative potential in the plane of adsorption, the less the amount of phosphate adsorption. Cation characteristics which determine the closeness of the cation from the surface are valency and the degree of the hydration. This may explain our finding that more P was extracted by sodium bicarbonate and sodium hydroxide when the more loosely bound Na ions were added compared to the addition of Ca ions. This observation is consistent with the results of the soil anion resin extractable P (as highlighted below) where rapid decline of P in solution was observed in the presence of Ca2+ and Cl− ions. This is supported by the increase in anion resin extractable P in the treatments when Na2CO3 was added. For Ca-saturated soils either by CaCl2 or CaCO3, an increase in concentration always produced an increase in phosphate partitioning into the solid phase as HCl fraction.
The supply of H+ is a driving force for the dissolution of PR, along with the removal of the dissolution reaction products Ca2+, H2PO4− and F− from the site of dissolution (Khasawneh and Doll 1978Khasawneh, F.E.; Doll, E.C. 1978. The use of phosphate rock for direct application to soils. Advances in Agronomy 30: 159–206.). The PR dissolution releases stoichiometric quantities of calcium ions into soil solution, which remain in the soil as exchangeable Ca (Khasawneh and Doll 1978Khasawneh, F.E.; Doll, E.C. 1978. The use of phosphate rock for direct application to soils. Advances in Agronomy 30: 159–206.). The presence of CaCO3has been shown to inhibit the dissolution of PR (Mackay and Syers, 1986MacKay, A.D.; Syers, J.K. 1986. Effect of phosphate, calcium, and pH on the dissolution of a phosphate rock in soil. Fertilizer Research 10: 175-184.) and this is due to both a Ca-common ion effect and an increase in pH caused by carbonate dissolution. Bolan and Hedley (1990)Bolan, N.S.; Hedley, M.J. 1990. Dissolution of phosphate rocks in soils. 2. Effect of pH on the dissolution and plant availability of phosphate rock in soil with pH dependent charge. Fertilizer Research 24: 125 134. reported increases in the dissolution of North Carolina phosphate rock, Jordan phosphate rock and Nauru phosphate rock in allophanic soils in New Zealand, when the soil pH decreased from 6.5 to 3.9.
Resin extractable P
The effect of added salts on anion resin-extractable P was less pronounced and in the opposite direction to that on sequential P (Figure 6). Values of resin P were highly correlated with the NaOH P fraction after addition of CaCO3, NaCl and CaCl2 (r2= 0.97, 0.99, 0.83 respectively), except when Na2CO3was added (r2 = 0.03). The trend was similar for the highly correlated regression analysis of resin P with HCl P after addition of CaCO3, NaCl and CaCl2 (r2 = 0.99, 0.97, 0.69 respectively) was also observed. There was a small increase in anionic resin P as Na2CO3 concentration increased, but decreased after 50 mmolc kg−1 (Figure 6). The decreasing resin extractable P declined as concentrations of NaCl, CaCl2, and CaCO3 increased suggesting that dissolution of PR decreased as the concentration of chloride and calcium increased..
Alternatively, this might suggest that secondary reactions were occurring between the soil and P released from PR. Consistent with this, the amount of P extracted with 0.5 M NaOH decreased and 1M HCl increased as the concentration of NaCl, Na2CO3, CaCl2 and CaCO3increased (Figure 2A and B) and accounted for the dissolved and residual PR remaining in the soil, respectively. However, calcium and chloride ions were the main influence on P recovery in the resin system.
CaCO3 inhibits the dissolution of PR (Mackay and Syers, 1986MacKay, A.D.; Syers, J.K. 1986. Effect of phosphate, calcium, and pH on the dissolution of a phosphate rock in soil. Fertilizer Research 10: 175-184.) due to both a Ca-common ion effect and an increase in pH caused by carbonate dissolution. The effect of suspension pH on PR dissolution supports the hypothesis of P precipitation induced by soluble Ca-ions. For Na-saturated soils (Na2CO3), an increase in pH always produced a decrease in phosphate partitioning into the solid phase. The increase in the soil solution P (Pi) that occurred after addition of Na2CO3 can be explained by assuming that Pi is controlled to a larger extent by the surface negative charge than by precipitation reactions. Thus, the increase in negative charge that occurs after addition of Na2CO3 would restrict P adsorption.
Assuming that P is preferentially adsorbed by sites with the more positive potential, as proposed by Barrow and Ellis (1986)Barrow, N.J.; Ellis, A.S. 1986. Testing a mechanistic model. II. The effect of pH on fluoride retention by a soil. Journal of Soil Science 37: 287-293., P adsorption would be restricted by a decrease in the positive surface potential that occurs after the addition of salts. However, the diminished PR dissolution with CaCO3 can be attributed to precipitation phenomena in which the suspension pH and the Ca solution influence the PR dissolution. When the Ca concentration in the solution is increased as well as higher pH values with CaCO3, the solubility of Ca-P mineral phases (PR) diminishes and precipitation phenomena increase distinctly, thus inhibiting PR dissolution.
To demonstrate the inhibitory effect of Ca on PR dissolution, a mixed resin system (AER+CER) was used for P extraction in treatments with calcium carbonate or calcium chloride. The use of AER+CER increased the resin extractable P in all the treatments in comparison to AER-P. The increase was highest when MPR was applied in combination with CaCO3 (Figure 7). In the AER+CER extraction system, the removal of calcium reduces the supply of Ca-ions in solution coming either from MPR and CaCO3 solubilization or Ca2+ exchange, thus reducing precipitation processes. The average increase in AER+CER-P due to incorporation of CaCO3 was 73 % (high rate of 80 mmolc kg−1) and 42 % (low rate of 50 mmolc kg−1) relative to no Ca. However, treatments that received MPR plus CaCl2 showed similar trends of reduced P extraction as with AER-P but with a different magnitude (Figure 7).
Mixed resin system (AER+CER) increased extractable P by 39 % (50 mmolckg−1) and 65 % (80 mmolc kg−1) over anion resin system (AER) when CaCl2 was added to MPR. The P extractability trend for the anion resin system was PR + CaCO3 > PR + CaCl2, while the mixed resin system (AER+CER) also followed a similar pattern but with a different magnitude ((AER+CER) > AER) (Figure 6). Resins generally function as sinks for ions in the medium; the acid cation exchange resins used here have a greater affinity for Ca ions compared with H counter ion initially present in the resin. PR dissolution is controlled largely by the size of the sink for solution Ca (Robinson et al., 1992bRobinson, J.S.; Syers, J.K.; Bolan, N.S. 1992b. Importance of proton supply and calcium sink size in the dissolution of phosphate rock materials of different reactivity in soil. Journal of Soil Science 43: 447-459.). As the proportion of Ca adsorbed by the cation exchange resin increased more PR dissolved releasing more P into the solution. This suggests that the effect of CaCO3 on PR dissolution is likely to depend on the Ca-sink size of the soil. The inclusion of the cation-anion resin system increased the Ca-sink size thus promoting MPR dissolution. This is a possible phenomenon/mechanism of increased P extraction where the mixed resin system was used. Tunesi et al. (1999)Tunesi, S.; Poggi, V.; Gessa, C. 1999. Phosphate adsorption and precipitation in calcareous soils: the role of calcium ions in solution and carbonate minerals. Nutrient Cycling in Agroecosystems 53: 219–227 in supernatant experiments, where soil minerals were removed to reduce the supply of Ca-ions in solution coming either from CaCO3 solubilization or Ca2+exchange, significantly reduced P precipitation processes.
Unlike NaOH P concentrations, (a measure of P strongly adsorbed onto Fe, Al oxides and kaolinitic clay), resin P concentrations, a measure of weakly adsorbed P exchangeable by HCO3− in the resin, was lower in all the treatments ( Table 2).
This is probably because the addition of CaCl2 lowered the soil pH, which generally increases the P sorption capacity of the soil (Mackay and Syers, 1986MacKay, A.D.; Syers, J.K. 1986. Effect of phosphate, calcium, and pH on the dissolution of a phosphate rock in soil. Fertilizer Research 10: 175-184.). Low pH and increased P-fixation will promote more PR dissolution, and therefore more P is in Al and Fe-P form which is not extracted with substantially increased efficiency by anion exchange resin. . This is consistent with the increased NaOH Pi extraction where either NaCl or CaCl2 was applied (Table 2).
Additionally, the above results are a phenomenon of the resin-soil system, specifically the accompanying anions in the Ca and Na treatments. In this scenario, the generally lower P extracted by both AER and AER+CER where CaCl2 and NaCl was applied could be attributed to a Cl effect. The strongly basic anion-exchange resins of the type used in this study show a specific and strong preference for Cl ions over orthophosphate ions (Christensen and Posner, 1980Christensen, H.H.; Posner, A.M. 1980. The interaction of phosphate with an anion exchange resin. Journal of Soil Science 31: 447–455.). These basic resins in the presence of high affinity counter ions (Cl−) are ineffective sinks for use in P studies. Savini et al. (2006)Savini, I.; Smithson, P.C.; Karanja, N.K. 2006. Effects of added biomass, soil pH and calcium on the solubility of Minjingu phosphate rock in a Kenyan Oxisol. Archives of Agronomy and Soil Science 52: 19-36. in an attempt to separate the relative contribution of Ca versus Cl effects on resin P extraction, have also shown that bicarbonate saturated anion resin extracted more P from TSP treated with Na2CO3 and CaCO3 than where NaCl, and CaCl was applied. They explained this in terms of the high concentration of complementary anions (chloride (Cl−) and the carbonate (CO3−)) inhibiting efficient extraction of soil P by the resin system.
Unlike the Cl−ion, the CO3−ion overcomes the complementary anion effect to some extent because CO3 is not conserved in solution. The carbonate ion is an ampholytic anion and may hydrolyze as shown below:
CO32– + 2H2O → H2CO3 + 2OH–
The considerable rise in pH for the lime-treated soils shows that the above route of reaction occurs. Conversely, at lower pH as in the Cl−-treated soil, more P is in Al and Fe-P form which is not extracted with substantially increased efficiency by the resin system. This was supported by the increase in the NaOH P fraction where Cl− was applied.
Effect of cations and anions on resin extractable P
Combined CERs/AERs are becoming increasingly popular as multi-element extractants for soils. Resins have the advantage of more closely simulating ion uptake characteristics of plant roots and correlations of resin extractable elements in soil with plant growth have generally been superior to those obtained using other chemical extractants (Skogley et al., 1990Skogley, E.O.; Georgins, S.J.; Yang, J.E.; Schaff, B.E. 1990. The phytoavailability soil test-P. Communication in Soil Science & Plant Analysis 21: 1229-1243.; Qain et al., 1992Qain, P.Y.; Schoenau, J.J.; Huang, W.Z. 1992. Use ion exchange membranes in routine soil testing. Communication in Soil Science & Plant Analysis 23: 1791-1804.). Addition of Na salts gave a similar pattern of anion resin extractable P for NaCl + TSP, Na2CO3 + MPR and Na2CO3+ TSP although with a different magnitude (Figure 8A).
− Effect of added salt on Minjingu phosphate rock and Triple Super phosphate anion resin P (A) Na2CO3 and NaCl (B) CaCO3 and CaCl2 (C) salt concentration.
The trend of anion resin extractable P after addition of Ca salts was CaCO3 + TSP > CaCl2 + TSP > CaCO3+MPR > CaCl2 + MPR (Figure 8B). Values of anion resin P were highly correlated with the amounts of Ca in the soil solution determined after application of the salts (r2 = 0.94). When values for the treatments with salts containing carbonate (CaCO3 and Na2CO3) were dropped from the regression analysis of bicarbonate P with resin P and NaOH P with resin P, the coefficients of correlation increased from 0.40 to 0.56 and 0.54 to 0.92 respectively. A similar (p < 0.05) increase in the coefficients of correlation was observed with the removal of salts containing sodium from the regression analysis (0.4 to 0.62 and 0.5 to 0.99 for bicarbonate P with resin P and NaOH P with resin P respectively). However, the increase in the correlation coefficients was more significant after exclusion of the treatment values with sodium rather than carbonate.
Cation characteristics which determine the closeness of surface approach are valence and degree of hydration. Replacement of Ca by the more loosely bound Na ion would, therefore, lead to desorption of P. Interestingly, soil incubated with PR+Na2CO3 increased (p< 0.05) resin-Pi concentrations for both MPR and TSP (Figure 8C). The magnitude of the increase in resin-Pi concentration due to the addition of MPR was similar due to the TSP addition to all the NaCO3 concentration (Figure 8C). However, low extractability of MPR resin P was observed when calcium chloride, calcium carbonate or sodium chloride was added. The resin-Pi concentrations showed a decrease with the increase in the application rates of CaCl2 and NaCl for MPR, but the rate of decrease was higher for the CaCl2 treatment than for the NaCl treatment (Figure 8C). Therefore, a determining role in the dissolution of phosphate rock is played by calcium ions made available either from the exchange complex or from the PR/calcite dissolution. We suggest that the high correlation (r2 = 0.99) found between AER + CER extractable P and the CaCO3 concentration in soil might be due mainly to the contribution of this solid phase in maintaining levels of exchangeable calcium sufficient to inhibit MPR dissolution or induce P precipitation.
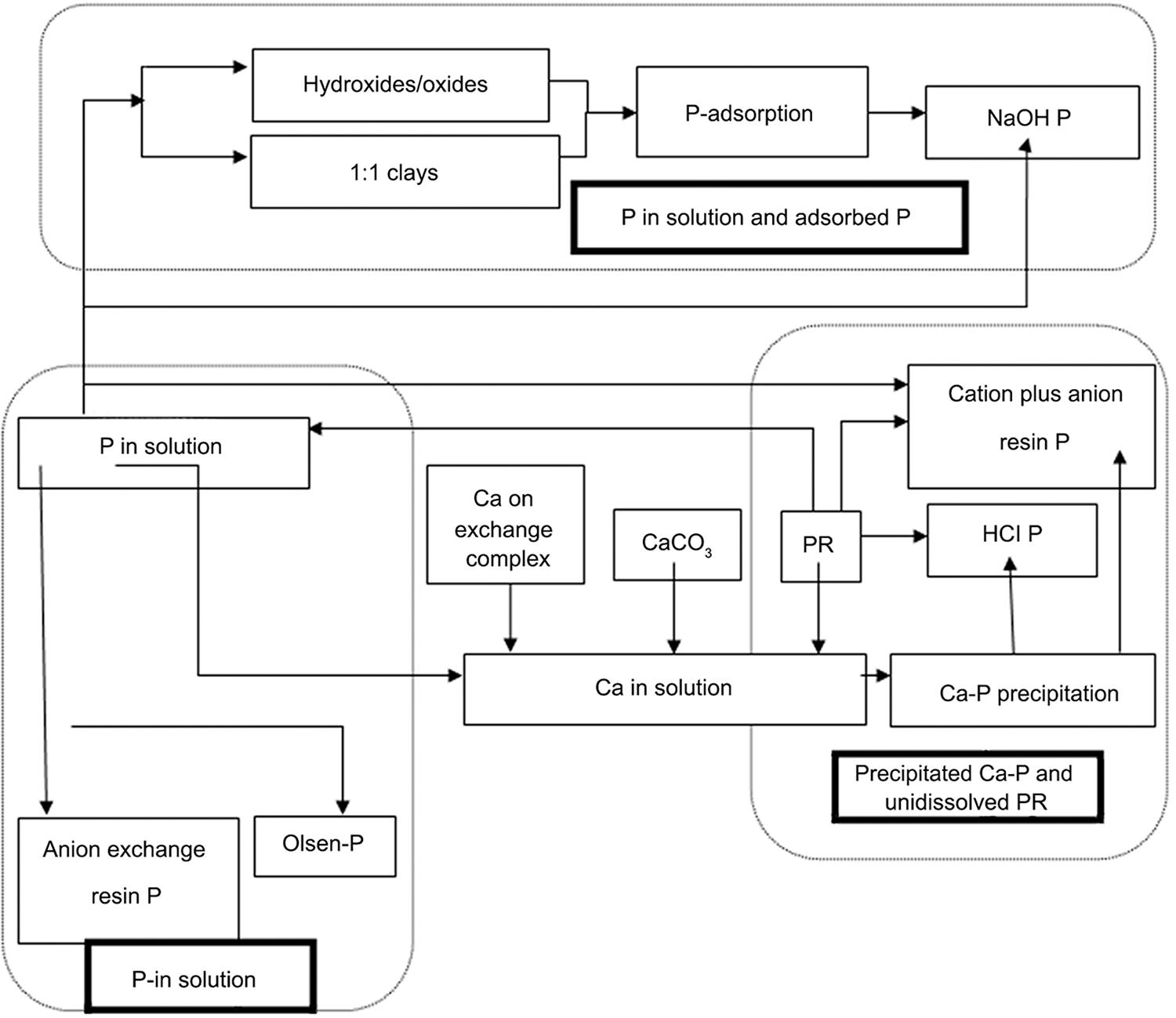
The contribution of several chemical processes to the partitioning of PR between solid and liquid phases can be summarized by: (i) adsorption on mineral components, such as clays, and hydroxides; and (ii) precipitation from solution, when solubility products for phosphate solid phases are exceeded by increasing concentrations of exchangeable cations such as Ca-ions; this process is strongly influenced by pH. The relationships proposed to describe how added Ca salts partitions PR into different P fractions (P in solution, Ca-precipitate and undissolved PR) and the estimation level of different P fractions in the soil-PR system are summarized in Figure 9.
Effect of NaCl-prewash on recovery of dissolved P
Syers et al. (1973)Syers, J.K.; Harris, R.F.; Armstrong, D.E. 1973. Phosphate chemistry in lake sediments. Journal of Environmental Quality 2: 1-14. have suggested that 1M NaCl extraction should be used prior to alkaline extractants in sequential P fractionation schemes. The 1M NaCl prewash removes exchangeable Ca thereby preventing the formation of Ca (OH)2 in the 0.5 M NaOH extracts, which could readsorb P or coprecipitate with extracted soil Pi. The effect of prewashing the soils used in this study with 1 M NaCl on 0.5 M NaOH and 0.5 M NaHCO3 extractants on the measurement of PR dissolution is presented in Table 3. The calculated amount of PR dissolved with NaCl prewash was significantly affected only where CaCl2 was applied (Table 3). The subsequent increase in 0.5 M NaOH and decrease in 1M HCl extractable P apparently results from the decrease in ionic strength and the replacement of exchangeable Ca and Mg by Na.
The prewash with NaCl removes Ca and avoids the formation of Ca(OH)2during the NaOH extraction, a fact confirmed by the greater P recoveries by the NaOH solution compared to the P recoveries in the absence of a prewash. Non-significant increases in the NaOH-P recoveries, except where CaCl2 was added, suggests that P is unlikely to be adsorbed by either Ca(OH)2 or CaCO3 precipitates or that these precipitates did not form during the extraction of this soil. Further, in acidic soils it is likely that the levels of exchangeable Ca are low and thus 1M NaCl pre-extraction is not required for the measurement of PR dissolution by 0.5 M NaOH method.
The current study reaffirms the need to remove Ca when evaluating the PR dissolution through alkaline extractions. Precipitation and insolubilization processes become dominant under increasing concentrations of calcium ions released from the exchange complex or solubilized from either carbonate or PR phases. The calcium released governs the insolubilization of phosphates, due to the low solubility of Ca–P solid phases.
We can conclude that concentration and identity of cations and anions in the soil solution may affect soil PR reactions and thus P availability. If solution mediated processes predominate in PR treated soils, agronomic practices which enrich the soil solution with reactants able to form Ca-complexes may be very important to increasing P availability to plants.
The interactive effects of PR, exchangeable cations and pH need to be taken into account when they are used in agriculture. The nature of the accompanying cation and pH were the main determinants of P recovery with sequential extraction.
Acknowledgements
This study was supported by a grant from the International Center for Tropical Agriculture - Tropical Soil Biology Fertility (CIAT-TSBF) and World Agroforestry Center.
References
- Alloush, G.A. 2003. Dissolution and effectiveness of phosphate rock in acidic soil amended with cattle manure. Plant and Soil 251: 37–46.
- Apthorp, J.N.; Hedley, M.J.; Tilman, R.W. 1987. The effect of nitrogen fertilizer form on the plant availability of phosphate from soil, phosphate rock and monocalcium phosphate. Fertilizer Research 12: 269-284.
- Baligar, V.C.; He, Z.L.; Martens, D.C.; Ritchey, K.D. 1997. Effect of phosphate rock, lime, and cellulose on ryegrass in an acidic soil. Plant and Soil 195: 129-136.
- Barrow, N.J.; Ellis, A.S. 1986. Testing a mechanistic model. II. The effect of pH on fluoride retention by a soil. Journal of Soil Science 37: 287-293.
- Bationo, A.; Mughogho, S.K.; Mokwunye, A.U. 1986. Agronomic evaluation of phosphate fertilizers in tropical Africa. p. 283-318. In: Mokwunye, A.U.; Vlek, P.L.G., eds. Management of nitrogen and phosphorus fertilizers in sub-Saharan Africa. Martinus Nijhoff, Dordrecht, The Netherlands.
- Bationo A., E. Ayuk, D. Ballo, and M. Koné. 1997. “Agronomic and Economic Evaluation of Telemsi Phosphate Rock in Different Agroecological Zones of Mali.” Nutrient Cycling in Agroecosystems, 48:179-189.
- Bolan, N.S.; Hedley, M.J. 1990. Dissolution of phosphate rocks in soils. 2. Effect of pH on the dissolution and plant availability of phosphate rock in soil with pH dependent charge. Fertilizer Research 24: 125 134.
- Bolan, N.S.; White, R.E.; Hedley, M.J. 1990. A review of the use of phosphate rocks as fertilizers for direct application in Australia and New Zealand. Australian Journal of Experimental Agriculture 30: 297-313.
- Bolland, M.D.A.; Gilkes, R.J. 1997. The agronomic effectiveness of reactive phosphate rocks, 2. Effect of phosphate rock reactivity. Australian Journal of Experimental Agriculture 37: 937-946.
- Bowden, J.W.; Posner, A.M.; Quirk, J.P. 1980. Adsorption and charging phenomena in variable charge soils. p. 147-166. In: Theng, B.K.G., ed. Soils with variable charge. New Zealand Society of Soil Science, Lower Hutt, New Zealand.
- Braun, A.R.; Smaling, E.M.A.; Muchugu, E.I.; Shepherd, K.D.; Corbett, J.D., eds. 1997. Maintenance and Improvement of Soil Productivity in the Highlands of Ethiopia, Kenya, Madagascar and Uganda: An Inventory of Spatial and Non-Spatial Survey and Research Data on Natural Resources and Land Productivity; African Highlands Initiative. International Centre for Research in Agroforestry, Nairobi, Kenya. (AHI Technical Report Series, 6).
- Chang, S.C.; Jackson, M.L. 1957. Fractionation of soil phosphorus. Soil Science 84: 133-144.
- Chapman, H.D. 1965. Cation exchange capacity. p. 891-901. In: Black, C.A.; Evans, D.D.; Ensminger, L.E.; White, J.L.; Clark, F.E., eds. Methods of soil analysis. Part 2. American Society of Agronomy, Madison, WI, USA. (Agronomy Monograph, 9).
- Chien, S.H. 1978. Interpretation of Bray I-extractable phosphorus from acid soils treated with phosphate rocks. Soil Science 126: 34-39.
- Chien, S.H.; Adams, F.; Khasawneh, F.E.; Henao, O. 1987. Effects of combinations of triple superphosphate and a reactive phosphate rock on yield and phosphorus uptake by corn. Soil Science Society of America Journal 51: 1656-1658.
- Chien, S.H.; Clayton, W.R.; McClellan, G.M. 1980a. Kinetic dissolution of phosphate rocks in soils. Soil Science Society of America Journal 44: 260-264.
- Chien, S.H.; Leon, L.A.; Tejeda, H. 1980b. Dissolution of North Carolina phosphate rock in acid Colombian soils as related to soil properties. Soil Science Society of America Journal 44: 1267-1271.
- Christensen, H.H.; Posner, A.M. 1980. The interaction of phosphate with an anion exchange resin. Journal of Soil Science 31: 447–455.
- Cross, A.F.; Schlesinger, W.H. 1995. A literature review and evaluation of the Hedley fractionation: applications to the biogeochemical cycles of soil phosphorus in natural ecosystems. Geoderma 64: 197–214.
- Curtin, D.; Syers, J. K.; Sraillie, G.W. 1987. The importance of exchangeable cations and resin-sink characteristics in the release of soil phosphorus. Journal of Soil Science 38: 711-716.
- Dewis, J.; Freitas, 1989. Physical and chemical methods of soil analysis. FAO, Rome, Italy. (FAO Soils Bulletin, 10).
- Engelstad, O.P.; Jugsujinda, A.; De Datta, S.K. 1974. Response by flooded rice to phosphate rocks varying in citrate solubility. Soil Science Society of America Proceedings 38: 524-529.
- Ernani, P.R.; Barber, S.A. 1995. Phosphorus availability in a low pH highly weathered soil as affected by added salts. Ciência Rural 25: 219-222.
- Fox, R.L.; Kamprath E.J. 1970. Phosphate sorption isotherms for evaluating the phosphate requirements of soil. Soil Science Society of America Proceedings 34: 902-907.
- Hartemink, A.E.; Buresh, R.J.; Jama, B.; Janssen, B.H. 1996. Soil nitrate and water dynamics in sesbania fallows, weed fallows and maize. Soil Science Society of America Journal 6: 568−574.
- He, Z.L.; Baligar, V.C.; Martens, D.C.; Ritchey, K.D.; Kemper, W.D. 1996b. Factors affecting phosphate rock dissolution in an acid soil amended with liming materials and cellulose. Soil Science Society of America Journal 60: 1596-1601.
- He, Z.L.; Baligar, V.C.; Martens, D.C.; Ritchey, K.D.; Kemper, W.D. 1996a. Kinetics of phosphate rock dissolution in an acid soil amended with liming materials and cellulose. Soil Science Society of America Journal 60: 1589-1595.
- Hedley, M.J.; Stewart, W.B.; Chauhan, B.S. 1982. Changes in inorganic and organic soil phosphorus fractions induced by cultivation practices and by laboratory incubations. Soil Science Society of America Journal 46: 970–976.
- Ivarsson, K. 1990. The long-term soil fertility experiments in southern Sweden. IV. Changes in inorganic and organic soil after a pot trial. Acta Agriculturae Scandinavic 40: 205–215.
- Kanabo, I.; Gilkes, R.J. 1987. The role of soil pH in the dissolution of phosphate rock fertilizers. Fertilizer Research 12: 165-174.
- Khasawneh, F.E.; Doll, E.C. 1978. The use of phosphate rock for direct application to soils. Advances in Agronomy 30: 159–206.
- Kihara J.; Martius, C.; Bationo, A.; Thuitaa M.; Lesueur, D.; Herrmann L.; Amelung, W.; Vlek, P.L.G. 2012 Soil aggregation and total diversity of bacteria and fungi in various tillage systems of sub-humid and semi-arid Kenya. Applied Soil Ecology 58: 12–20.
- Kirk, G.J.D.; Nye, P.H. 1986. A simple model for predicting the rates of dissolution of sparingly soluble calcium phosphates in soil. III. A predictive model for regularly distributed particles. Journal of Soil Science 37: 511–524.
- Lindsay, W.L.; Vlek, P.L.G. 1977. Phosphate minerals. p. 639-672. In: Dixon, J.B.; Weed. S.B., eds. Minerals in soil environments. Soil Science Society of America, Madison, WI, USA.
- MacKay, A.D.; Syers, J.K.; Tillman, R.W.; Gregg, P.E.H. 1986. A simple model to describe the dissolution of phosphate rock in soils. Soil Science Society of America Journal 50: 291-296.
- MacKay, A.D.; Syers, J.K. 1986. Effect of phosphate, calcium, and pH on the dissolution of a phosphate rock in soil. Fertilizer Research 10: 175-184.
- McLean, E.O. 1965. Soil pH and lime requirement. p. 199-223. In: Black, C.A.; Evans, D.P.; White, J.L.; Ensminger, L.E; Clark, F.E., eds. Methods of soil analysis. Part 2. American Society of Agronomy, Madison, WI, USA.
- Menon, R.G.; Hammond, L.L,; Sissingh, H.A. 1989. Determination of plant available phosphorus by the iron hydroxide-impregnated filter paper (Pi) soil test. Soil Science Society of America Journal 53: 110–115.
- Murphy, J.; Riley, J.P. 1962. A modified single solution method for the determination of phosphorus in natural waters. Analytica Chimica Acta 27: 31-36.
- Mutuo, P.K.; Smithson, P.C.; Buresh, R.J.; Okalebo, R.J. 1999. Comparison of phosphate rock and triple superphosphate on a phosphorus-deficient Kenyan soil. Communication in Soil Science & Plant Analysis 30: 1091–1103.
- Naidu, R.; Syers, J.K.; Kirkman, J.H. 1990. Effect of liming on phosphate sorption by acid soils. Journal of Soil Science 41: 165–175.
- Nelson, D.W.; Sommers, L.E.; 1985. Total carbon, organic carbon and organic. p. 539-579. In:Page, A.L., ed. Methods of soil analysis. Part 2. Chemical and microbiological properties. Soil Science Society of America, Madison, WI, USA.
- Okalebo, J.R.; Gathua, K.W.; Woomer, P.L. 1993. Laboratory methods of plant and soil analysis: a working manual. KARI, SSEA, TSBF, UNESCO-ROSTA, Nairobi, Kenya.
- Olsen, S.R.; Cole, C.V.; Watanabe, F.S.; Dean, L.A. 1954. Estimation of vailable phosphorus in soils by extraction with sodium bicarbonate. USDA, Washingtom, DC, USA. (Circular, 939).
- Pissarides, A.J.; Stewart, W.B.; Reenie, D.A. 1968. Influence of cation saturation on phosphorus adsorption by selected clay minerals. Canadian Journal of Soil Science 48: 151-157.
- Qain, P.Y.; Schoenau, J.J.; Huang, W.Z. 1992. Use ion exchange membranes in routine soil testing. Communication in Soil Science & Plant Analysis 23: 1791-1804.
- Robinson, J.S.; Syers, J.K. 1990. A critical evaluation of factors influencing the dissolution of Gafsa phosphate rock. Journal of Soil Science 41: 597-605.
- Robinson, J.S.; Syers, J.K.; Bolan, N.S. 1992b. Importance of proton supply and calcium sink size in the dissolution of phosphate rock materials of different reactivity in soil. Journal of Soil Science 43: 447-459.
- Sample, E.C.; Soper, R. J.; Racz, J.G. 1980. Reactions of phosphate fertilizers in soils. p. 263-310. In: Khasawneh, F.E.; Sample, E.C.; Kamprath, E.J., eds. The role of phosphorus in agriculture. ASA/CSSA/SSSA, Madison, WI, USA.
- Sanyal, S.K.; Datta, S.K.D. 1991. Chemistry of phosphorous transformations in soil. Advances in Soil Science 16: 1–94.
- Savini, I.; Smithson, P.C.; Karanja, N.K. 2006. Effects of added biomass, soil pH and calcium on the solubility of Minjingu phosphate rock in a Kenyan Oxisol. Archives of Agronomy and Soil Science 52: 19-36.
- Schmidt, J.P.; Buol, S.W.; Kamprath, E.J. 1996. Soil phosphorus dynamics during seventeen years of continuous cultivation: Fractionation analysis. Soil Science Society of America Journal 60: 1168–1172.
- Sharpley, A.N. 1991. Soil phosphorus extracted by iron-aluminum line oxide-impregnated filter paper. Soil Science Society of America Journal 55: 1038–1041.
- Sibbesen, E. 1978. An investigation of the anion-exchange resin method for soil phosphate extraction. Plant and Soil 50: 305–321.
- Skogley, E.O.; Georgins, S.J.; Yang, J.E.; Schaff, B.E. 1990. The phytoavailability soil test-P. Communication in Soil Science & Plant Analysis 21: 1229-1243.
- Sposito, G. 2008. The Surface Chemistry of Soils. Oxford University Press, New York, NY, USA.
- Stoorvogel, J.J.; Smaling, E.M.A,; Jansen, B.H. 1993. Calculating soil nutrient balances in Africa at different scales. I. Supra-National scale. Fertilizer Research 35: 227-235.
- Syers, J.K.; Harris, R.F.; Armstrong, D.E. 1973. Phosphate chemistry in lake sediments. Journal of Environmental Quality 2: 1-14.
- Tiessen, H.; Stewart, J.W.B.; Cole, C. V.1984. Pathways of Phosphorus Transformations in Soils of Differing Pedogenesis. Soil Science Society of America Journal 48: 853-858.
- Tiessen, H.; Moir, J.O. 1993. Characterization of available phosphorus by sequential extraction. p. 76–86. In: Carter, M.R., ed. Soil sampling and methods of analysis. Lewis, Boca Raton, FL, USA.
- Tunesi, S.; Poggi, V.; Gessa, C. 1999. Phosphate adsorption and precipitation in calcareous soils: the role of calcium ions in solution and carbonate minerals. Nutrient Cycling in Agroecosystems 53: 219–227
- United States Department of Agriculture [USDA]. 1992. Soil Taxonomy. Washington, DC, USA. (Agriculture Handbook, 294).
- Walker, T.W.; Syers, J.K. 1976. The fate of phosphorus during pedogenesis. Geoderma 15: 1–19.
- Wiklander, L. 1964. Cation and anion exchange phenomena. p. 163-205. In: Bear, F.E., ed. Chemistry of the soil. Reinhold, New York, NY, USA.
- Wilson, M.A.; Ellis, B.G. 1984. Influence of calcium solution activity and surface area on the solubility of selected rock phosphates. Soil Science 138: 354–359.
- Zapata, F.; Zaharah, A.R. 2002. Phosphorus availability from phosphate rock and sewage sludge as influenced by the addition of water-soluble phosphate fertilizer. Nutrient Cycling in Agroecosystems 63: 43-48.
- Zee, S.E.A.T.M. van der; Fokkink, L.G.J.; Riemsdijk, W.H. van. 1987. A new technique for assessment of reversibly adsorbed phosphate. Soil Science Society of America Journal 51: 599–604.
Edited by
Publication Dates
-
Publication in this collection
Sep-Oct 2015
History
-
Received
20 Aug 2014 -
Accepted
23 Mar 2015