Abstracts
Tuberous Sclerosis Complex, also known as Epiloia or Bourneville-Pringle disease is an autosomal dominant neurocutaneous syndrome with variable clinical expression. It is a multisystem disorder that may be associated with hamartomas in multiple organs in an unpredictable manner. The dermatologist plays an essential role in the history of the disease, since skin manifestations represent the most prevalent clinical features, enabling early diagnosis and intervention in its natural course. This article aims to inform the scientific community about advances made in the study of genetics and molecular biology. Recent findings regarding stimulation of tumor growth have been changing the history of this condition, making therapeutic trials with topical and systemic drugs possible. Knowledge of these topics enables better management of the patients affected, since tissue replacement by tumors can result in significant morbidity and mortality.
Dermatology; Diagnosis; Neurology; Sirolimus; Tuberous sclerosis
A Esclerose Tuberosa, também conhecida como Epilóia ou Facomatose de Pringle-Bourneville, é uma síndrome neurocutânea de caráter autossômico dominante com expressões clínicas variadas. É uma doença multissistêmica que pode cursar com hamartomas em diversos órgãos, de forma imprevisível. O dermatologista tem papel essencial na história da doença, uma vez que as afecções cutâneas representam as mais prevalentes apresentações clínicas, possibilitando assim o diagnóstico precoce da síndrome e intervenção na sua evolução natural. O presente artigo tem o objetivo de atualizar a comunidade científica sobre avanços alcançados no estudo genético e biologia molecular. Recentes descobertas sobre o estímulo do crescimento tumoral vêm mudando a evolução desta patologia, possibilitando ensaios terapêuticos com drogas tópicas e sistêmicas. O conhecimento destes aspectos possibilita melhor condução dos pacientes acometidos, dado que a substituição tumoral dos diversos tecidos pode resultar em relevante morbidade e mortalidade.
Dermatologia; Diagnóstico; Esclerose tuberosa; Neurologia; Sirolimo
CONTINUED MEDICAL EDUCATION
Tuberous sclerosis complex* * Work conducted at the University Hospital of Brazilia - University of Brazilia (UNB-HUB) - Brazilia (DF), Brazil.
Esclerose tuberosa
Daniela Araujo RodriguesI; Ciro Martins GomesII; Izelda Maria Carvalho CostaIII
IMD, Specialist in Dermatology certified by the Brazilian Society of Dermatology - Preceptor of the Medical Residency Program in Dermatology, University Hospital of Brazilia - University of Brazilia (UnB-HUB) - Brazilia (DF), Brazil
IIResident in Dermatology, University Hospital of Brazilia - University of Brazilia (UnB-HUB) - Brazilia (DF), Brazil
IIIPhD in Dermatology, Federal University of Sao Paulo (UNIFESP) - Professor of Dermatology at the University of Brazilia (UnB), Coordinator of the outpatient department of pediatric dermatology, University Hospital of Brazilia - University of Brazilia (UnB-HUB) - Brazilia (DF), Brazil
Mailing address Mailing address: Daniela Araujo Rodrigues Hospital Universitário de Brasília SGAN 605, Av. L2 Norte 70910-900 Brasília, DF E-mail: danirodrigues@unb.br
ABSTRACT
Tuberous Sclerosis Complex, also known as Epiloia or Bourneville-Pringle disease is an autosomal dominant neurocutaneous syndrome with variable clinical expression. It is a multisystem disorder that may be associated with hamartomas in multiple organs in an unpredictable manner. The dermatologist plays an essential role in the history of the disease, since skin manifestations represent the most prevalent clinical features, enabling early diagnosis and intervention in its natural course. This article aims to inform the scientific community about advances made in the study of genetics and molecular biology. Recent findings regarding stimulation of tumor growth have been changing the history of this condition, making therapeutic trials with topical and systemic drugs possible. Knowledge of these topics enables better management of the patients affected, since tissue replacement by tumors can result in significant morbidity and mortality.
Keywords: Dermatology; Diagnosis; Neurology; Sirolimus; Tuberous sclerosis
RESUMO
A Esclerose Tuberosa, também conhecida como Epilóia ou Facomatose de Pringle-Bourneville, é uma síndrome neurocutânea de caráter autossômico dominante com expressões clínicas variadas. É uma doença multissistêmica que pode cursar com hamartomas em diversos órgãos, de forma imprevisível. O dermatologista tem papel essencial na história da doença, uma vez que as afecções cutâneas representam as mais prevalentes apresentações clínicas, possibilitando assim o diagnóstico precoce da síndrome e intervenção na sua evolução natural. O presente artigo tem o objetivo de atualizar a comunidade científica sobre avanços alcançados no estudo genético e biologia molecular. Recentes descobertas sobre o estímulo do crescimento tumoral vêm mudando a evolução desta patologia, possibilitando ensaios terapêuticos com drogas tópicas e sistêmicas. O conhecimento destes aspectos possibilita melhor condução dos pacientes acometidos, dado que a substituição tumoral dos diversos tecidos pode resultar em relevante morbidade e mortalidade.
Palavras-chave: Dermatologia; Diagnóstico; Esclerose tuberosa; Neurologia; Sirolimo
BACKGROUND
Tuberous sclerosis complex (TSC), also known as Epiloia or Bourneville-Pringle disease, is an autosomal dominant neurocutaneous syndrome with variable clinical expression. It is a multisystem disease that may be associated with hamartomas in various organs in an unpredictable manner. It most often affects the skin and central nervous system. 1
Centuries ago, isolated case reports described some of the various clinical manifestations resulting from a pathology which was then unknown. In the mid 19th century, Virchow and Von Recklinghausen identified hamartomas in the brain and heart, respectively. 2 The term ''Tuberous'' was coined after a postmortem examination of the brains of patients who had seizures and cognitive impairment, due to the aspect of the tumors found, which resembled tuberous vegetables such as potatoes. Only in the late 19th century were the findings concomitantly described by Bourneville, who first reported the discovery of a new syndrome.3
Although cutaneous manifestations had been previously described by other authors, they started to be related to the disease described by Bourneville only in the early 20th century. The first to recognize this relationship were Campbell, in 1905, and Vogt, three years later, when the relationship between mental retardation, epilepsy, and sebaceous adenoma was established, forming the triad characterizing TSC.4
TSC is a complex clinical entity made up of many clinical manifestations, which require attention from various medical specialties. The dermatologist plays an essential role in the history of the disease, since cutaneous involvement represents the most prevalent finding, enabling early diagnosis of the syndrome and intervention in its natural course.
After years of studies limited to several clinical aspects and certain genetic aspects of TSC, the era of molecular biology started. The new objects of interest are the signaling pathways of hamartoma growth. Great success in the recognition of proteins, enzymes and signals involved in the etiopathological process of the syndrome has recently been obtained. These findings are used to understand diseases that have uncontrolled growth of tumor cells as a common causal factor, such as TSC. The advances that have been made in the area are relevant, and today, two centuries after the first description of tubers by Virchow and Von Recklinghausen, it is possible to perform clinical trials of new drugs that can alter the course of this disease that causes considerable morbidity and mortality.5,6
EPIDEMIOLOGY
Studies assessing the incidence and prevalence of Tuberous Sclerosis Complex express different results. It is a disease with great phenotypic variability, which sometimes hinders its recognition.7 It affects 1 in 10,000 newborns, and the majority of patients are diagnosed with the disease in the first 15 months of life.6,8 About 25% of the individuals diagnosed after that age had previously showed characteristic clinical signs that were overlooked during a previous medical evaluation.4,8 With the advent of new techniques for genetic study and the definition of new clinical manifestations, we have an estimated prevalence of 1:6,000 persons in the general population.6 Both sexes are affected in a similar frequency, but women may show more prominent signs. There are no reports showing disproportionate involvement in a particular ethnic group. 9
The sign that most often leads to diagnosis of the syndrome is the presence of early-onset seizures. 6,8 Involvement of the skin and mucous membranes also helps with the diagnosis; however, the set of characteristic skin manifestations tends to appear at later stages. The incidence of disorders of certain organ systems is variable, but neurological and renal complications are the leading causes of mortality and morbidity.10
ETIOPATHOGENESIS
Tuberous sclerosis complex is an autosomal dominant disease with high penetrance. It is caused by inactivating mutations of TSC1 and TSC2 tumor suppressor genes, located on chromosomes 9q34 and 16p13.3, respectively.11 The TSC1 gene is responsible for encoding a protein called hamartin and TSC2 for encoding for tuberin. The hamartin-tuberin complex is an important inhibitor of tumor growth. Its absence triggers loss of inhibition on cell proliferation and migration.3
Numerous genetic changes are observed, such as deletions, insertions, nonsense, and missense mutations, basically involving all exons of the genes involved.6 In spite of verification of such changes, mutations are not identified in 15% of the cases.3 The familial forms of TSC arise from germline mutations and, although their transmission can be hereditary, 70% of the cases are a result of somatic mutations, especially when the mutant gene is TSC2. Somatic mutations are responsible for sporadic cases of the disease.6 The great variety of mutations and the possibility of involvement of different genes explain, in part, the phenotypic diversity of the disease. Another factor that may explain why clinical manifestations of TSC are unpredictable is the presence of mosaicism.12,13
Studies show that changes in TSC2 are five times more frequent than changes in TSC1 in sporadic cases. This relationship becomes 1:1 in cases of familial transmission.6 Changes in the TSC2 gene result in more severe pathology, when compared to TSC resulting from mutations in TSC1.14-16 Cases of familial transmission, which show higher frequency of changes in the TSC1 gene, result in mild to moderate disease, sometimes not meeting the various diagnostic criteria.16
For several years, the "two-hit" hypothesis, proposed by Knudson in the 70's, explained the etiopathogenesis of hamartoma formation in TSC. According to that hypothesis, patients with TSC would already present a defective tumor suppressor gene in one of the alleles of TSC1 or TSC2 during germ-cell division. This stage would be the "first hit". However, this previous change would not be enough to trigger phenotypic findings, since both alleles must be lost or deficient so that clinical manifestation occurs. Thus, a second change involving the normal allele and leading to loss of heterozygosity is necessary for the development of a hamartoma in TSC. This would be the "second hit", which occurs during somatic cell proliferation. Knudson believes that the presence of the anomalous allele creates genomic instability or induces epigenetic phenomena (genetic changes caused by non-mutational factors, such as DNA methylation), which trigger loss of heterozygosity and disrupt the function of the tumor suppressor gene. This genetic change would lead to absence of the hamartin-tuberin complex, resulting in uncontrolled cell growth.18,19 Some studies have recently challenged this hypothesis, due to the finding that tumors have been developed in spite of an intact normal allele.20
The mechanism through which these mutations lead to cellular hyperproliferation and appearance of numerous hamartomas anywhere in the human body is not fully understood. Some of the stages of this proliferation pathway have already been discovered. The cells of the human body continuously respond to a wide variety of integrated biological stimuli. Defects in this signaling mechanism result in proliferative diseases such as cancer and TSC. The TSC1 and TSC2 genes have an important role in regulating cell growth, via the phosphoinositide 3-kinase signaling pathway, inhibiting the mammalian target of rapamycin (mTOR). mTOR is a major sensor of energy and nutrient availability, thus regulating cell growth, differentiation and proliferation.5,21 As mentioned above, the TSC1 gene encodes for a 130Kda protein known as hamartin and TSC2 encodes for a 200Kda protein called tuberin.22 The hamartin-tuberin complex forms a heterodimer that inhibits the signaling protein, known as "RAS homolog enriched in brain" (Rheb).6 mTOR, in turn, following stimulation by Rheb, regulates the phosphorylation of ribosomal protein S6 kinase 1 and 2 (S6K1 and S6K2) isoforms and of the protein inhibiting translation initiation, 4E-BP1 (also known as PHAS-1).21,23-25 The Loss of inhibitory function of the hamartin-tuberin complex on Rheb allows mTOR to stimulate the S6K and 4E-BP1 proteins, leading to an increase in ribosomal biogenesis, with increased production of proteins, which ultimately culminates in uncontrolled cell proliferation and tissue growth, with the emergence of tumors observed in tuberous sclerosis.26-29
The role of TSC1 and TSC2 in regulating biological processes is not yet fully understood, and some of its properties are the subject of considerable interest. One example is the finding that the hamartintuberin complex acts in the biology of the cell cycle by regulating cyclin-dependent kinase inhibitor p27, playing a major role in the stages of cell differentiation.30 Loss of inhibition of the mTOR signaling pathway also has an important role in the insulin-dependent cellular metabolism. Activation of mTOR induces endoplasmic reticulum stress, resulting in severe insulin and IGF1 resistance, besides making the cell more susceptible to apoptosis.5
Other clinical manifestations can be explained by genetic changes different from alterations in the TSC1 and TSC2. The best example is the concomitant occurrence of TSC and polycystic kidney disease, an important cause of end-stage renal disease in these patients. They may be considered contiguous gene syndromes, due to the proximity of the two mutant genes responsible for the two diseases. The concomitant occurrence of both diseases results from two large deletions in the TSC2 gene, leading to mutation, by contiguity, of the PKD1 gene, which is responsible for polycystic kidney disease. Both genes are located on chromosome 16.31-34
CLINICAL ASPECTS
Tuberous sclerosis presents clinical manifestations resulting from the formation of hamartomas in various organs. These lesions are dependent on the different types of genetic changes found in the pathophysiology of the disease, a fact which contributes to the existence of various forms of phenotypic TSC.35
Skin Manifestations
According to a study that examines clinical signs on the skin based on age group, skin clinical manifestations represent the most frequent findings in TSC, although some patients do not show skin involvement. 36 Among these skin clinical manifestations, the most prevalent is the presence of hypomelanotic macules (Figure 1). Hypochromic/achromic macules are found in 90 to 98% of the patients, especially before puberty and may be the only manifestation in children. 3,36 Parents report the presence of such macules at birth or in early life, affecting mainly the trunk and limbs, usually sparing the face. These macules tend to increase in number and size throug-hout life, becoming less prominent in adults, when they become more pigmented, and may even disappear. When the achromic macules affect the scalp, there may be poliosis.36,37
The most characteristic hypochromic/achromic macules are those that have the shape of leaves, rounded at one end and tapered at the other, and therefore, called "ash-leaves'' (Figure 2).36 They can also be rounded, "confetti-like", in 28% of the cases. These may be multiple, with 1-2mm in diameter, symmetrically affecting the distal part of the limbs, along their entire circumference (Figure 3). The "confetti-like" hypomelanosis is often underdiagnosed due to lack of routine use of ultraviolet light, which shows dyschromia more easily. A histopathological evaluation of achromic macules shows a normal number of melanocytes, but that melanosomes are reduced in number, size, and melanization. There may be "café-au-lait" macules, but some studies consider their prevalence the same as that found in the general population.36
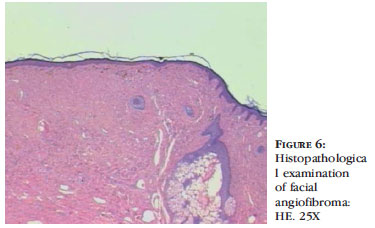
Angiofibromas are characterized by skin-colored to violaceous papules, depending on the higher proportion of fibrous or angiomatous tissue, respectively. They are present in 80% of affected children who are older than 5.3 They primarily affect the nasolabial folds, cheeks and chin, bilaterally and symmetrically, sparing the upper lip (Figures 4, 5 and 6). When they occur unilaterally, there may be mosaicism.36-38 Clinical observations report excessive malar erythema before the onset of angiofibromas.36
Shagreen patch is a skin-colored to brown fibrotic plaque which appears at around age 3, but may already be present at birth (Figure 7).37,38 It occurs in 54% of affected children over 5 years old, usually at puberty, and almost always occurs in the lower dorsal surface, with multiple satellite papules. 3 Skin-colored papules grouped at the characteristic site of the plaque may be considered equivalent.36-38
Forehead fibrous plaques, although not as prevalent, are considered pathognomonic for TSC. They present as a unilateral plaque with a fibrotic aspect in the frontal region and may be skin-colored or brown (Figure 8).38
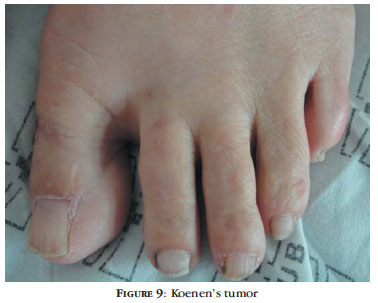
Periungual fibroma or Koenen's tumor is one of the last changes to occur in TSC and its incidence tends to increase with age (Figure 9).39 Tumors tend to be multiple over the years, affecting mainly females, especially the nails of the toes. In some cases, there is only a longitudinal depression in the nail, corresponding to the fibroma still in the nail matrix (Figure 10) or to the remnant of the tumor, after being excised by the patient himself.36
ORAL CAVITY
The teeth and gum tissue may be affected in TSC. Gingival fibromas affect about 36% of adults with TSC and are rarely found in children younger than 11 years old. They are usually detected in the anterior gingiva and may result in flesh-colored or erythematous lesions. 36,40 Enamel pits have also been described, but they are not considered pathognomonic for TSC and are often underdiagnosed on examination.41 Therefore, a detailed assessment of the oral cavity is crucial, including examination of the gingiva under dentures, for the detection of these changes.
RENAL MANIFESTATIONS
Renal changes are the main cause of morbidity and mortality in TSC. Among them, the most common
are renal angiomyolipomas, which appear before adolescence and are present in 93% of the cases.3 They are usually bilateral and asymptomatic. They may be the cause of intense hematuria, which is more common in tumors larger than 4 cm, and may lead to loss of renal function. Their growth is slow, but in rare cases, their size may increase by up to 4 cm every 2 years. 37,38
Concomitant polycystic kidney disease occurs due to the proximity of the polycystic kidney disease 1 (PKD1) gene in relation to TSC2 gene, both located on chromosome 16. This disease affects mainly children, resulting in progressive loss of renal function and may culminate in chronic renal failure. 37,38
Patients with TSC have an estimated incidence of renal cell carcinoma close to that of the general population. However, these tumors develop early, increasing morbidity and mortality of the patients affected by TSC.1
PULMONARY MANIFESTATIONS
In TSC, the expression of lung disease seriously affects the function of the organ, due to replacement of alveolar tissue by numerous cysts and proliferation of smooth muscle, called pulmonary lymphangioleimyomatosis (LAM).42 It occurs in 1 to 3% of the cases, preferably in women during premenopause. Pulmonary involvement is rare in males. This predilection for females may be explained by the regulatory action of estrogen on cellular signaling pathways involved in TSC and on the migration of deficient cells of TSC2. The symptoms are cough and progressive dyspnea, and may also include hemoptysis and pneumothorax. Besides presenting therapeutic difficulty, the evolution of LAM is progressive, which offers a guarded prognosis, and may lead to respiratory failure.3
Heart changes
Rhabdomyoma is the most common cardiac tumor in TSC. Since it is more prevalent early in life, it is the most frequent clinical change in fetuses and newborns.43 They are usually located in the wall of the ventricles and, depending on size, number and location, can result in cardiomegaly, heart murmurs, altered blood flow, arrhythmias, non-immune hydrops fetalis and death. They usually regress completely in the first years of life, due to lack of maternal hormone stimulation.44 Despite the variety of symptoms resulting from rhabdomyoma, most cases are asymptomatic.43
In a recent study, Adriaensen et al. evaluated chest CT of 55 patients with TSC and found foci of fat attenuation in 64% of the cases. The authors suggest that such findings may be part of the characteristics of TSC; however, its true clinical significance remains uncertain. 45
Neurological Changes
The most common neurological clinical manifestations in TSC are epilepsy, cognitive delay and autism. 46 Seizures are routine in the childhood of patients with TSC, and infantile spasm is considered to be the most commonly diagnosed subtype in the first year of life.37 Epilepsy may be present in up to 90% of the cases. Its early onset is associated with its refractoriness and more severe cognitive delay.46
Epileptogenesis in TSC can be explained by decreased neuronal inhibition secondary to molecular alterations in GABA receptors, which are present in giant cells and dysplastic neurons. GABAergic deficiency may explain early onset and severity of seizures.3 An explanation suggested for refractory seizures in TSC, after immunohistochemical evaluation of surgical specimens from epileptic patients with TSC, is the overexpression of P-glycoprotein, encoded by MDR1 gene, in the epileptogenic tissue. This glycoprotein could be responsible for clearance of antiepileptic drugs, and would be increased in these cases, leading to poor therapeutic response. 47
In a study carried out by means of serial analysis of interictal electroencephalogram (EEG) records of patients with TSC, it was found that patients with more than three interictal epileptogenic foci tend to have poorer cognitive development. It was also shown that the earlier the onset of seizures, the greater the number of interictal epileptogenic foci.48
An important datum in clinical practice is that West syndrome, which comprises spasm-type seizures, delayed psychomotor development and hypsarrhythmia in the electroencephalogram, is the most frequent epileptic encephalopathy in the first year of life and that, after severe neonatal asphyxia and congenital brain malformations, TSC is the most frequent cause of West syndrome. Hence the need to diagnose these pathologies, which can be associated, since 30% of children under 1 year of age with TSC and epilepsy will develop West syndrome.49
Neuroimaging studies are altered in 90 to 95% of cases of TSC, with the description of tubers in the cerebral cortex, subependymal nodules in the walls of the lateral ventricles and subependymal giant cell tumors. 36 Serial studies that have evaluated the relationship between imaging and clinical manifestations of TSC have identified that a greater number of tubers, their bilateralism and location in the temporal lobe are directly related to greater severity of neurological involvement.50,51
On histopathological examination, cortical tubers in the brain show absence of normal laminated architecture. There is also presence of large cells resembling astrocytes but positive for neuronal and glial markers. Subependymal nodules present vascular stroma and astrocyte-like cells.36
Ocular changes
Calcified retinal astrocytic hamartoma can be present in 50% of patients.37,52 They tend to be bilateral and become evident at the age of 2. They are asymptomatic in most cases, in which case they do not affect the optic nerve or macula. Patients may also present hypochromic macule in the retina, corresponding to skin macules.3
Liver changes
Multiple hepatic angiomyolipomas have been reported in rare cases of TSC, and this prevalence may be the result of underdiagnosis, for they are usually asymptomatic. Ultrasonographic evaluation of patients with TSC shows higher prevalence of hepatic hamartomas in adulthood (23-45%). There is some predilection for females.3
Diagnosis
Diagnosis of tuberous sclerosis is initially based on clinical findings. The disorder should be considered in cases of children with seizures and hypomelanotic macules.
Due to the clinical variability of TSC, clinical and radiological diagnostic criteria were revised in 2000. Diagnosis can be definitive, probable or possible, depending on the number of major and minor criteria present (Table 1).37,53 Definitive diagnosis is considered when there is presence of at least two major criteria or one major and two minor criteria. Probable diagnosis is considered when there is presence of one major and one minor criterion. Possible diagnosis refers to the presence of one major criterion or two or more minor criteria.36,37,53 Some considerations regarding TSC diagnostic criteria should be stressed. When cortical dysplasia and cerebral white matter radial migration occur together, they shall be considered as only one criterion. When LAM and renal angiomyolipomas occur concomitantly, other criteria for TSC must be present, before confirming the diagnosis. It is suggested that hamartomatous rectal polyps, multiple renal cysts and nonrenal hamartomas be histologically confirmed. Identification through imaging examinations is sufficient for the diagnosis of cerebral white matter radial migration and bone cysts.53
Earlier at the onset of clinical manifestations of TSC, some additional tests should be performed in order to confirm the diagnosis and to elucidate the causes of symptoms. Thus, before the diagnostic possibility of TSC, cranial computed tomography (CT) or magnetic resonance imaging (MRI) must be requested; electroencephalogram must be requested in case of seizures; renal ultrasound must be performed in all suspected patients; echocardiography, electrocardiogram, ophthalmoscopy and pulmonary CT must be requested for women; pulmonary function tests should be performed in women with respiratory symptoms; renal function tests should be requested in cases of children with polycystic kidneys and of adults with extensive renal involvement. Neuropsychomotor development and behavior should also be assessed in an attempt to establish the presence of autism, hyperactivity and mental retardation.37
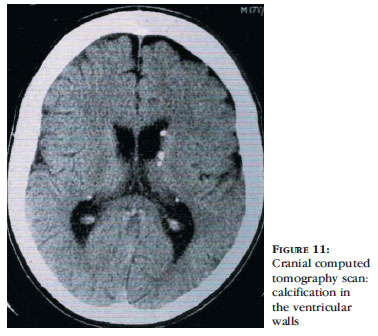
Echocardiography may be altered due to the presence of rhabdomyomas, even before neurological and skin lesions become noticeable.44 Cranial CT scanning shows subependymal nodules, often calcified, present in the walls of the lateral ventricles and which may be protruding into the ventricular cavity (Figure 11).3,36 For evaluation of cortical tubers, MRI of the brain, using FLAIR sequences, shows to be superior to other neuroimaging studies. With this method, tubers may be associated with white matter abnormalities, such as radial migration lines.36,37,52 Fetal MRI shows alterations from the 26th week of pregnancy, when there are tubers in the central nervous system.3
Although it is not part of the classic diagnostic criteria due to its low availability, a genetic study may be asked for confirmation and detection of the mutation trig-gering TSC. This test may also be useful in evaluating and counseling healthy relatives of patients with TSC.
MONITORING
Brain MRI or cranial CT scan should be performed every 1 to 3 years in children and less frequently in adults. Early diagnosis of intracranial changes enables complete removal of tumors, when indicated.37
Chest CT scan and pulmonary function tests should be performed every 6 to 12 months in symptomatic women with pulmonary LAM.37 Renal ultrasound should be obtained every 1 to 3 years in adolescents and adults for monitoring the emergence of renal angiomyolipomas.3,37
GENETIC COUNSELING
The active search for cases of TSC in undiagnosed relatives, due to a less prominent clinical manifestation, is important for the establishment of early treatment of possible complications caused by tumors and also for raising awareness of the possible transmissibility of TSC to future generations. It is known that a patient with TSC has a 50% chance of having a child affected by the disease, and the risk of a healthy couple who had a child with TSC to have another child with the disease is 2%.37
Medical evaluation of healthy family members who are planning to have children should be performed by means of a dermatological examination, due to the delayed onset of facial angiofibromas and ungual fibromas in some cases, in addition to renal ultrasonography and cranial CT scan.36
Genetic testing, as mentioned above, help clarify mutations responsible for cases of TSC, in addition to evaluating changes in healthy relatives so that the risk of another child being born with the disease is established. In spite of this, about 2% of healthy parents of children with TSC present gonadal mosaicism. Thus, such mutations would not be diagnosed during genetic evaluation, which means that the risk of the next child to be affected by TSC would remain.3
Genetic evaluation is not routinely indicated for monitoring of patients affected by TSC. Before an uncertain clinical and radiological diagnosis, the need for evaluation of family members and prenatal diagnosis of the disease, molecular genetic testing helps the physician in clinical practice. According to the Tuberous Sclerosis Alliance, genetic testing for Tuberous sclerosis complex can be performed by means of polymerase chain reaction, DNA sequencing and TSC1 and TSC2 dosage. 54,55
TREATMENT
Treatment of TSC consists in addressing the symptoms caused by hamartomas and in prophylactic measures to prevent loss of function of the affected organ. Since it is a systemic disease, a multidisciplinary approach is mandatory.
Facial angiofibromas may be removed by dermabrasion, surgical excision, electrocautery and argon laser therapy, and their recurrence is common.36,37 Fibrous tumors are best treated with CO2 laser.37 Due to the recurrent and progressive growth of facial tumors, surgical treatment may be delayed until after adolescence, when their growth is at maximum peak.36 Ungual fibromas that present frequent bleeding or are painful may be surgically excised, treated with electrocautery or argon laser, but recurrence is not uncommon.36,37
Renal angiomyolipomas that cause persistent hematuria should undergo arterial embolization. In tumors larger than 4 cm, there should be close monitoring, with evaluation of the need for prophylactic embolization, since these are the ones most likely to cause severe hematuria.37 This procedure should also be performed in centrally located tumors, when surgical treatment poses greater risks. Angiogenesis inhibitors may be used to prevent the development of angiomyolipomas and may improve the prognosis of TSC. 3
Cardiac rhabdomyomas should be surgically removed if symptomatic. Cardiac arrhythmias are treated clinically.37
Anticonvulsants should be used in patients with epilepsy, and the most effective drug is vigabatrin. In cases of infantile spasms, the use of corticotropin or prednisolone is recommended.37 In patients with TSC and refractory epilepsy, surgical resection of epileptogenic foci has decreased drug resistance in these patients by 57%. Surgery can reduce the deterioration of cognitive function and behavioral disorders.37 In case of subependymal astrocytoma, surgical resection is preferred over radiotherapy, since response to the latter is unsatisfactory and since it increases the occurrence of glioblastomas. 37
After the discovery that regulation of the mTOR pathway is associated with the development of tumors in TSC, some promising studies have been receiving great attention. Interferon-gamma and -alpha interact with the mTOR pathway, leading to deactivation of 4E-BP1, which could be very helpful in the treatment of TSC.3
Inhibitors of the mTOR pathway, such as rapamycin, also known as sirolimus, have an immunosuppressive and antiproliferative action. They are responsible for normalizing the functioning of this pathway in cells deficient in TSC1 or TSC2. This drug is effective in reducing the volume of tumors in patients with TSC, such as renal angiomyolipoma, subependymal giant cell astrocytoma and sporadic LAM. Patients with mutations in TSC2 presented learning improvement and patients with LAM showed recovery of the pulmonary function after using rapamycin. Despite the satisfactory therapeutic effect in the treatment of renal angiomyolipomas, recurrence of uncontrolled tumor growth after discontinuation of the drug was observed.56 Topical therapy with rapamycin was shown to be feasible and safe in a clinical study conducted with patients with psoriasis.57 The real role of this route of administration is promising, due to the possibility of reduced side effects and greater effect on the skin. However, there is no consistent scientific evidence to date.58
It is believed that, in the future, gene therapy will make it possible to treat mutations that cause TSC through reintroduction of defective genes, leading to induction of apoptosis of mutated cells, or through reintroduction of the lost gene, enabling the cell to present normal phenotype and avoiding the clinical changes of TSC. 59,60

Received on 24.01.2011.
Approved by the Advisory Board and accepted for publication on 24.03.2011.
Conflict of interest: None
Financial funding: None
QUESTIONS

- 1. Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87-105.
- 2. Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Mosby: Elsevier; 2007.
- 3. Curatolo P, Bombardieri R, Jozwiak S. Tuberous Sclerosis. Lancet. 2008;372:657-68.
- 4. Lendvay TS, Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol. 2003;169:1635-42.
- 5. Ozcan U, Ozcan L, Yilmaz E, Düvel K, Sahin M, Manning BD, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541-51.
- 6. Napolioni V, Curatolo P. Genetics and molecular biology of tuberous sclerosis complex. Curr Genomics. 2008;9:475-87.
- 7. Roach ES, DiMario FJ, Kandt RS, Northrup H. Tuberous Sclerosis Consensus Conference: recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol. 1999;14:401-7.
- 8. Devlin LA, Shepherd CH, Crawford H, Morrison PJ. Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Dev Med Child Neurol. 2006;48:495-9.
- 9. Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ. Fitzpatrick's Dermatology in General Medicine. 7th ed. New York: Mc Graw-Hill; 2007.
- 10. O'Callaghan FJ, Noakes MJ, Martyn CN, Osborne JP. An epidemiological study of renal pathology in tuberous sclerosis complex. BJU Int. 2004;94:853-7.
- 11. Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87-96.
- 12. Yates JR, van Bakel I, Sepp T, Payne SJ, Webb DW, Nevin NC, et al. Female germline mosaicism in tuberous sclerosis confirmed by molecular genetic analysis. Hum Mol Genet. 1997;6:2265-9.
- 13. Rose VM, Au KS, Pollom G, Roach ES, Prashner HR, Northrup H. Germ-line mosaicism in tuberous sclerosis: how common? Am J Hum Genet. 1999;64:986-92.
- 14. Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64-80.
- 15. Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999;64:1305-15.
- 16. Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotypephenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13:731-41.
- 17. Au KS, Hebert AA, Roach ES, Northrup H. Complete inactivation of the TSC2 gene leads to formation of hamartomas. Am J Hum Genet. 1999;65:1790-5.
- 18. Green AJ, Smith M, Yates JR. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet. 1994;6:193-6.
- 19. Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, Hornigold N, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59:400-6.
- 20. Han S, Santos TM, Puga A, Roy J, Thiele EA, McCollin M, et al. Phosphorylation of tuberin as a novel mechanism for somatic inactivation of the tuberous sclerosis complex proteins in brain lesions. Cancer Res. 2004;64:812-6.
- 21. Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPaseactivating protein complex toward Rheb. Curr Biol. 2003;13:1259-68.
- 22. Nellist M, Goedbloed MA, Halley DJ. Regulation of tuberous sclerosis complex (TSC) function by 14-3-3 proteins. Biochem Soc Trans. 2003;31:587-91.
- 23. Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472-87.
- 24. Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457-66.
- 25. Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5:566-71.
- 26. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19-24.
- 27. Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14 Spec No. 2:R251-8.
- 28. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596-603.
- 29. Dann SG, Thomas G. The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett. 2006;580:2821-9.
- 30. Rosner M, Freilinger A, Hengstschläger M. Akt regulates nuclear/cytoplasmic localization of tuberin. Oncogene. 2007;26:521-31.
- 31. Gomez MR. Tuberous Sclerosis. 2nd ed. New York: Raven Press, 1988.
- 32. Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979-87.
- 33. Brasier JL, Henske EP. Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J Clin Invest. 1997;99:194-9.
- 34. Stillwell TJ, Gomez MR, Kelalis PP. Renal lesions in tuberous sclerosis. J Urol. 1987;138:477-81.
- 35. Lendvay TS, Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol. 2003;169:1635-42.
- 36. Webb DW, Clarke A, Fryer A, Osborne JP. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol. 1996;135:1-5.
- 37. Yates JR. Tuberous sclerosis. Eur J Hum Genet. 2006;14:1065-73.
- 38. Webb DW, Clarke A, Fryer A, Osborne JP. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol. 1996;135:1-5.
- 39. Aldrich CS, Hong CH, Groves L, Olsen C, Moss J, Darling TN. Acral lesions in tuberous sclerosis complex: insights into pathogenesis. J Am Acad Dermatol. 2010;63:244-51.
- 40. Martelli H, Lima LS, Bonan PR, Coletta RD. Oral manifestations leading to the diagnosis of familial tuberous sclerosis. Indian J Dent Res. 2010;21:138-40.
- 41. Pereira ALM. Esclerose tuberosa: relato de caso. An Bras Dermatol. 1998;73:431-4.
- 42. Park HY, Nam HS, Chung MP, Jeong SH, Kim YJ, Cha SI, et al. A nationwide survey of lymphangioleiomyomatosis in Korea: recent increase in newly diagnosed patients. J Korean Med Sci. 2010;25:1182-6.
- 43. Quek SC, Yip W, Quek ST, Chang SK, Wong ML, Low PS. Cardiac manifestations in tuberous sclerosis: a 10-year review. J Paediatr Child Health. 1998;34:283-7.
- 44. Yinon Y, Chitayat D, Blaser S, Seed M, Amsalem H, Yoo SJ, et al. Fetal cardiac tumors: a single-center experience of 40 cases. Prenat Diagn. 2010;30:941-9.
- 45. Adriaensen ME, Schaefer-Prokop CM, Duyndam DA, Zonnenberg BA, Prokop M. Fatty foci in the myocardium in patients with tuberous sclerosis complex: common finding at CT. Radiology. 2009;253:359-63.
- 46. Samir H, Ghaffar HA, Nasr M. Seizures and intellectual outcome: Clinico-radiological study of 30 Egyptian cases of tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15:131-7.
- 47. Lazarowski A, Lubieniecki F, Camarero S, Pomata H, Bartuluchi M, Sevlever G, et al. Multidrug resistance proteins in tuberous sclerosis and refractory epilepsy. Pediatr Neurol. 2004;30:102-6.
- 48. Jansen FE, van Huffelen AC, Bourez-Swart M, van Nieuwenhuizen O. Consistent localization of interictal epileptiform activity on EEGs of patients with tuberous sclerosis complex. Epilepsia. 2005;46:415-9.
- 49. Garg RK. Infantile spasms. Indian Pediatr. 1997;34:220-6.
- 50. Chou IJ, Lin KL, Wong AM, Wang HS, Chou ML, Hung PC, et al. Neuroimaging correlation with neurological severity in tuberous sclerosis complex. Eur J Paediatr Neurol. 2008;12:108-12.
- 51. Zaroff CM, Barr WB, Carlson C, LaJoie J, Madhavan D, Miles DK, et al. Mental retardation and relation to seizure and tuber burden in tuberous sclerosis complex. Seizure. 2006;15:558-62.
- 52. Rosser T, Panigrahy A, McClintock W. The diverse clinical manifestations of tuberous sclerosis complex: a review. Semin Pediatr Neurol. 2006;13:27-36.
- 53. Hyman MH, Whitemore VH. National Institute of Health Consensus Conference: tuberous sclerosis complex. Arch Neurol. 2000;57:662-5.
- 54. Tsalliance.org [Internet]. Silver Spring: Tuberous Sclerosis Alliance. [cited 2011 Feb 24]. Available from: http://www.tsalliance.org/pages.aspx?content=588
- 55. Athenadiagnostics.com [Internet]. Worcester: Athena diagnostics. [Cited 2011 Feb 24]. Available from: http://www.athenadiagnostics.com/content/test-catalog/
- 56. Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140-51.
- 57. Ormerod AD, Shah SA, Copeland P, Omar G, Winfield A. Treatment of psoriasis with topical sirolimus: preclinical development and a randomized, double-blind trial. Br J Dermatol. 2005;152:758-64.
- 58. Haemel AK, O'Brian AL, Teng JM. Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol. 2010;146:715-8.
- 59. Ward LS. Entendendo o Processo Molecular da Tumorigênese. Arq Bras Endocrinol Metab. 2002;46:351-60.
- 60. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416-22.
Publication Dates
-
Publication in this collection
03 May 2012 -
Date of issue
Apr 2012
History
-
Received
24 Jan 2011 -
Accepted
24 Mar 2011