Abstract
The field of mitochondrial DNA (mtDNA) replication has been experiencing incredible progress in recent years, and yet little is certain about the mechanism(s) used by animal cells to replicate this plasmid-like genome. The long-standing strand-displacement model of mammalian mtDNA replication (for which single-stranded DNA intermediates are a hallmark) has been intensively challenged by a new set of data, which suggests that replication proceeds via coupled leading-and lagging-strand synthesis (resembling bacterial genome replication) and/or via long stretches of RNA intermediates laid on the mtDNA lagging-strand (the so called RITOLS). The set of proteins required for mtDNA replication is small and includes the catalytic and accessory subunits of DNA polymerase y, the mtDNA helicase Twinkle, the mitochondrial single-stranded DNA-binding protein, and the mitochondrial RNA polymerase (which most likely functions as the mtDNA primase). Mutations in the genes coding for the first three proteins are associated with human diseases and premature aging, justifying the research interest in the genetic, biochemical and structural properties of the mtDNA replication machinery. Here we summarize these properties and discuss the current models of mtDNA replication in animal cells.
DNA replication; mitochondria; mtSSB; pol γ; Twinkle
REVIEW ARTICLE
Replicating animal mitochondrial DNA
Emily A. McKinney; Marcos T. Oliveira
Institute of Biomedical Technology, University of Tampere, Tampere, Finland
Send correspondence to Send correspondence to Marcos T. Oliveira Institute of Biomedical Technology, University of Tampere, Biokatu 6, 33014 Tampere, Finland. E-mail: marcos.oliveira@uta.fi
ABSTRACT
The field of mitochondrial DNA (mtDNA) replication has been experiencing incredible progress in recent years, and yet little is certain about the mechanism(s) used by animal cells to replicate this plasmid-like genome. The long-standing strand-displacement model of mammalian mtDNA replication (for which single-stranded DNA intermediates are a hallmark) has been intensively challenged by a new set of data, which suggests that replication proceeds via coupled leading-and lagging-strand synthesis (resembling bacterial genome replication) and/or via long stretches of RNA intermediates laid on the mtDNA lagging-strand (the so called RITOLS). The set of proteins required for mtDNA replication is small and includes the catalytic and accessory subunits of DNA polymerase y, the mtDNA helicase Twinkle, the mitochondrial single-stranded DNA-binding protein, and the mitochondrial RNA polymerase (which most likely functions as the mtDNA primase). Mutations in the genes coding for the first three proteins are associated with human diseases and premature aging, justifying the research interest in the genetic, biochemical and structural properties of the mtDNA replication machinery. Here we summarize these properties and discuss the current models of mtDNA replication in animal cells.
Keywords: DNA replication, mitochondria, mtSSB, pol y, Twinkle.
Introduction
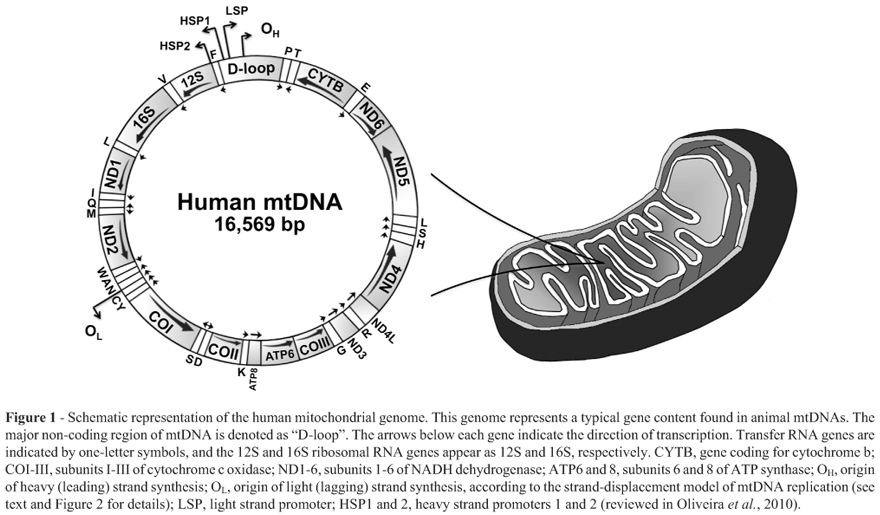
For decades, the mitochondrial genome was thought to be a less important plasmid-like DNA, which contained residual genetic information from the ancestral a-proteobacterium that eventually became the mitochondrion, determining one of the most important endosymbiotic events in the evolutionary history of eukaryotes. We now know that an intact and functional mitochondrial genome (Figure 1) is required for normal assembly and operation of the complexes involved in mitochondrial oxidative phosphorylation (OXPHOS), and therefore, for the bulk of ATP production (Scheffler, 2008). Mitochondria also perform a myriad of functions in eukaryotic cells that are interconnected to ATP synthesis, such as autophagy, calcium signaling, apoptosis, and production of intermediates in a variety of biosynthetic pathways (reviewed in Nunnari and Suomalainen, 2012). For the correct performance of this interconnected system, and thus, for proper physiological homeostasis/adaptability, cells depend on the qualitative and quantitative status of the mitochondrial genome. It is, therefore, easy to understand how point mutations, deletions or rearrangements in the mitochondrial DNA (mtDNA) molecule, and up-or down-regulation of mtDNA copy number in a particular tissue/organ can influence the organism's phenotype by promoting disease states or selectively (dis)advantageous conditions.
In this review, we focus on the recent discoveries in mtDNA replication in animal cells. Researchers in the last two decades have studied the subject on two fronts: 1) investigating the composition of mtDNA replication intermediates in vivo and in cells in culture; and 2) investigating the biochemical and physical properties of the proteins involved in mtDNA maintenance. These studies have shown that the mtDNA transactions are more diverse and complex than previously thought (for more details on these transactions, see Oliveira et al., 2010), revealing new mechanisms of genome replication with implications for the understanding of human mitochondrial disorders and the evolution of animal mitochondria.
Modes of mtDNA Replication in Animal Cells: One, Two or Three Ways of Duplicating a Genome
Historically, replication of mammalian mitochondrial genomes was studied using cesium chloride-purified mtDNA, visualized by electron microscopy (Robberson et al., 1972). In combination with S1 protection, radiolabeling studies and ligation-mediated PCR (Bogenhagen et al., 1979; Nass, 1980a,b; Tapper and Clayton, 1981; Kang et al., 1997), this extensive body of work has generated a model of mtDNA replication that has been standing for over 35 years, referred to as the strand-displacement model (Bogenhagen and Clayton, 2003a). In this model (Figure 2A), the synthesis of the leading strand (known as the heavy [H] strand in vertebrate mtDNA because of its guanine-rich composition) initiates within the non-coding Dloop region at a site designated as OH, and proceeds continuously. As leading strand synthesis continues, replication intermediates accumulate a progressively larger displaced parental H strand, maintained in a single-stranded form. At about two thirds of the distance around the genome, the origin of the lagging strand (or light [L] strand) synthesis OL is exposed, allowing replication of the lagging strand to be initiated and synthesis to proceed continuously. The overall process does not involve the formation of Okazaki fragments; in summary, replication is unidirectional, continuous, asymmetric and asynchronous. Recently, this model has been updated by data from atomic force microscopy, which revealed alternative L strand origins (Brown et al., 2005). Moreover, biochemical data have shown that the mitochondrial RNA polymerase can in fact make an RNA primer specifically at OL, which can be used by DNA polymerase γ (see next section for proteins of the mtDNA replisome) to initiate synthesis of the L strand (Fuste et al., 2010). In vitro, the mitochondrial RNA polymerase is also able to prime the displaced leading strand at multiple points during synthesis of the new leading strand (see more details in the next section); the gaps between primers can then be filled by DNA polymerase γ (Wanrooij et al., 2008). Although the authors concluded that this data supports the revised strand-displacement model, it also suggests the presence of Okazaki-like fragments during synthesis of the mtDNA lagging strand, a class of replication intermediates that has to date not been observed in the mitochondrion.
Since the year 2000, two other models of mtDNA replication have been reported in the literature, challenging the validity of the long-standing strand-displacement model. Studies using two-dimensional agarose gel electrophoresis (2DAGE) and a variety of nucleic acid-modifying enzymes have revealed evidence for a new class of replication intermediates: extensive segments of RNA incorporated into the newly synthesized lagging strand, hybridized to the template leading strand (Yang et al., 2002; Yasukawa et al., 2006). This model implies strand-coupled replication proceeding unidirectionally from the D-loop region, with the RNA replication intermediates subsequently being maturated into DNA. The RITOLS (RNA Incorporated ThroughOut the Lagging Strand) model of mtDNA replication was then born (Figure 2B). More recently, this model has gained tremendous strength due to the observation via transmission electron microscopy and immunopurification using antibodies specific to RNA/DNA hybrids that replicating mammalian mtDNA molecules are essentially duplex with extensive RNA tracts present in one strand (Pohjoismaki et al., 2010). Furthermore, interstrand crosslinking experiments and in organello DNA synthesis indicate that the RNA/DNA hybrids are present in vivo (Reyes et al., 2013), and are not an artifact of the mitochondrial nucleic acid preparations, as previously suggested by Bogenhagen and Clayton (2003a,b).
The second model, referred to as the strand-coupled model, was proposed by the same groups of researchers using 2DAGE (Holt et al., 2000); this replication mode, however, appears to occur only when cells are recovering from ethidium bromide-induced mtDNA depletion. The hallmark of the model (Figure 2C) is the presence of fully double-stranded DNA theta-like replication intermediates indicative of coupled leading and lagging DNA strand synthesis, as it is found in bacterial DNA replication. Interestingly, in this case, replication starts at a broad initiation zone, in a region containing the genes cytb, nad5 and nad6 of the mammalian mitochondrial genome, and proceeds bidirectionally until finally arresting at the D-loop region (Bowmaker et al., 2003). Although the 2DAGE data is substantial, this model invokes the existence of Okazaki fragments as replication intermediates and two polymerases working in a single replisome, which have yet to be shown for complete validation of the model.
In our opinion, the strand-coupled and the RITOLS models most likely describe interconnected events, as parts of the same process: the RNA hybridized to the old leading strand, during synthesis of the new leading strand, may be processed and serve as multiple priming sites for the DNA synthesis of the new lagging strand. Speculatively, the efficient action of the mitochondrial forms of DNA ligase III, Flap endonuclease 1 and Dna2 would then promote RNA removal and nick sealing, completing the maturation of the newly replicated mtDNA (reviewed in Holt, 2009). Evidence for this interplay between strand-coupled and RITOLS has been reported by Wanrooij et al. (2007), who expressed catalytically mutant forms of the DNA polymerase y and the mtDNA helicase Twinkle in human cells in culture (see next section for proteins of the mtDNA replisome). Cells expressing Twinkle mutants accumulated double-stranded mtDNA replication intermediates (the main feature of the strand-coupled model) with loss of RNA associated with the lagging strand (the hallmark of the RITOLS model), compared to control cells. This indicates an increased rate of initiation of lagging strand synthesis and/or RNA-DNA maturation relative to the rate of replication fork movement (or synthesis of the leading strand), consistent with the role of Twinkle in unwinding double-stranded DNA. On the other hand, expression of pol γ mutants induced replication stalling but maintained the RNA replication intermediates, primarily because of delayed lagging-strand DNA synthesis or its maturation.
Finding a relation between the strand-displacement model and the strand-coupled/RITOLS models has been more difficult, primarily because it appears that the regions of the mammalian mtDNA that are essentially singlestranded according to the former model, are the same regions which contain the RNA replication intermediates in accordance to the latter models. Remarkably, Pohjoismäki et al. (2010) showed loss of these RNA molecules when they followed the protocol for preparation of mitochondrial nucleic acids used to describe the strand-displacement model. The single-strandness of the long-standing stranddisplacement model of mtDNA replication might, in the end, just be a technical artifact. In addition, in the race to find the "right" mode of mtDNA replication, research on the strand-coupled/RITOLS models has accumulated a significant amount of supportive data, "passing" most (if not all) of the tests imposed by critics (Bogenhagen and Clayton, 2003a,b). The research has also gone beyond mammalian organisms, showing the possible conservation of mechanisms in birds (Reyes et al., 2005) and in Drosophila melanogaster (Joers and Jacobs, 2013). Unfortunately for the field, researchers of the strand-displacement model have not maintained the same pace of investigations using in vivo data, which might compromise the credibility of the oldest model.
The mtDNA Replisome: The Tools to Use When Duplicating a Genome
All proteins involved in animal mtDNA maintenance are encoded by nuclear genes, and to date, only three of these proteins have been identified working at the mtDNA replication fork (Figure 3). Ahead of the fork, the replicative helicase Twinkle translocates on one DNA strand (5' to 3' direction), unwinding double-stranded DNA (dsDNA) into single-stranded DNA (ssDNA). DNA polymerase γ (pol γ) can then perform DNA synthesis per se, using as a template the ssDNA released by Twinkle. This ssDNA is also bound to the mitochondrial single-stranded DNA-binding protein (mtSSB), which protects it from nucleolysis, without any associated catalytic activity. Furthermore, replication does not appear to be primed by RNA derived from a dedicated primase (see discussion below), but instead by extension of processed RNA transcripts laid down by the mitochondrial RNA polymerase (mtRNApol) (Reyes et al., 2013). [Technically, there is no experimental evidence showing that mtRNApol is part of the mtDNA replisome and moves along with the replication fork, but we have included this enzyme here due to its increasingly important roles in mtDNA replication]. Interestingly, sequence and structural analyses indicate that the mtDNA replisome is a "chimeric machinery", consisting of proteins of different evolutionary origins. Twinkle, the catalytic subunit of pol γ (pol γ-α) and mtRNApol share ancestry with the gp4 primase-helicase, the gp5 DNA polymerase and the gp1 RNA polymerase from the bacteriophage T7, respectively, whereas mtSSB is a homologue of the homotetrameric SSBs from eubacteria (Shutt and Gray, 2006a). The accessory subunit of pol γ (pol γ-β) appears exclusively in the metazoan lineage and has clearly evolved from the class II aminoacyl-tRNA synthetases of eubacteria (Fan et al., 1999, 2006). How these proteins of originally distinct functions have evolved to participate in mtDNA replication is still unclear, although the functional rewiring of ancestral proteins appears to have happened frequently in mitochondrial history (Shutt and Gray, 2006a; Camara et al., 2011).
Twinkle, pol γ-α and pol γ-β have gained a lot of interest recently because of the discovery that mutations in these genes are associated with the outcome of human diseases (Table 1), such as progressive external ophthalmoplegia (PEO), Alpers' syndrome, ataxia-neuropathy disorders, among others (reviewed in Euro et al., 2011; Martin-Negrier et al., 2011; Stumpf et al., 2013). The Human DNA Polymerase Gamma Mutation Database shows approximately 200 mutations in the gene coding for pol γ-α and 9 in the gene coding for pol γ-β (also known as POLG1 and POLG2 genes, respectively); these were found in patients with diagnosed mitochondrial disease or who presented symptoms suggestive of mitochondrial disease. Pol γ-α, which contains the enzyme's 5'-3' polymerase, 3'-5' exonuclease and 5' deoxyribose-5-phosphate lyase activities, is the most studied polypeptide of the mtDNA replisome at the clinical, biochemical and structural levels. The mutations found in POLG1, are nearly uniformly distributed along the length of the gene (Stumpf and Copeland, 2011), but they do tend to cluster within distinct functional modules in the tridimensional structure of the enzyme, at least for the mutations associated with the severe Alpers' syndrome (Euro et al., 2011). Interestingly, Alpers patients typically present compound heterozygosity, but there has not yet been a case in which a combination of two mutations from the same functional module was found. The understanding of the spatial orientation of mutated residues in pol γ-α structure allows, for the first time, that computational predictions can be made to support the pathogenic role of newly discovered pol γ variants (Euro et al., 2011).
In addition to its role in human diseases, pol γ has also recently been implicated in aging. A mouse model expressing a proofreading-deficient pol γ-α (the "mtDNA mutator" mouse), which has a D257A substitution in its exonuclease domain, showed increased mtDNA mutation rates and clear mitochondrial defects, as predicted (Trifunovic et al., 2004; Kujoth et al., 2005; Vermulst et al., 2007, 2008; Edgar and Trifunovic, 2009). Surprisingly, the most pronounced phenotypes of these mice were reduced longevity and premature aging, characterized by a loss of weight (reduction in subcutaneous fat content and in whole-body bone mineral density and content), kyphosis, alopecia, anemia, infertility, hearing loss and reduced hair density, detected at a young age (24-25 weeks). Although it appears that the mtDNA mutator mouse does not have increased levels of reactive oxygen species (ROS) due to its mitochondrial dysfunctions, this animal model clearly establishes the role of mtDNA mutations as a cause of aging and age-related diseases. Mitochondrial ROS are generated when the transport of electrons in the respiratory chain is interrupted (by chemical inhibition or mutations in subunits of the OXPHOS complexes), causing a leak of these electrons, which will consequently react directly with molecular oxygen, creating highly reactive molecules that have DNA, lipid and protein damaging properties. This chain of events is part of the foundation of the mitochondrial theory of aging; data from the mutator mouse suggests that this theory needs some reevaluation to accommodate the idea that mtDNA mutations might be involved with other aging mechanisms, and not with elevated ROS production (reviewed in Bratic and Larsson, 2013).
Mutations in the human Twinkle gene (C10orf2) were for the first time reported in 2001 as a cause of autosomal dominant PEO (Table 1), associated with multiple mtDNA deletions (Spelbrink et al., 2001). It was not until 2003-2004 that biochemical studies showed the NTPase, 5'-3' ssDNA translocase and dsDNA unwinding activities of the encoded enzyme, establishing it as the replicative mtDNA helicase (Korhonen et al., 2003, 2004). Combined with mutagenesis studies that demonstrated the roles of conserved residues in the Walker A and Walker B motifs and identified the arginine finger of Twinkle (Matsushima and Kaguni, 2007; Matsushima et al., 2008), the discovery of the abovementioned enzymatic activities related to helicase function is not as surprising as the fact that Twinkle does not contain primase activity. The sequence similarities between Twinkle and the bifunctional gp4 primase-helicase of bacteriophage T7, which suggest that these enzymes share ancestry, are most pronounced in the C-terminal helicase domain (Spelbrink et al., 2001). The N-terminal "primase"-like domain of the human Twinkle (and all other vertebrates) has diverged dramatically, losing nearly all conserved residues important for synthesis of RNA primers (Shutt and Gray, 2006b) and, most importantly, showing no detectable primase activity in vitro (Farge et al., 2008). Even the Drosophila melanogaster Twinkle, which unlike its vertebrate homologues, retained most of the conserved cysteines found in the primase domain of T7 gp4 (Shutt and Gray, 2006b), does not appear to need these residues during mtDNA replication in cultured S2 cells (Matsushima and Kaguni, 2009).
These observations give us the opportunity to speculate on the roles of the N-terminal "primase"-like domain of Twinkle and on the nature of the putative mtDNA primase. Because the N-terminus of Twinkle has the ability to bind ssDNA (Farge et al., 2008) and this property is required for the recently and unexpectedly identified strand annealing activity of the enzyme (Sen et al., 2012), Twinkle may play a role in recombination-mediated replication initiation, as found in the brain and heart of mammalian mitochondria (Pohjoismaki et al., 2009), or in replication fork regression during repair of damaged DNA replication forks (Cheok et al., 2005). Additionally, the N-terminus might be important for some of the putative roles of Twinkle as the initiator of mtDNA replication (Jemt et al., 2011; Milenkovic et al., 2013). Therefore, the enzyme responsible for making the RNA primers which will be used by pol γ during DNA synthesis appears to be the only DNA-dependent RNA polymerase found in the mitochondrion, mtRNApol. In vitro studies showed that mtRNApol can synthesize short RNA primers for DNA synthesis of mtDNA lagging strand (Wanrooij et al., 2008), but in vivo, evidence indicate that the RNA primers come from the processing of mature transcripts, including messenger and transfer RNAs, hybridized to the lagging-strand template (Reyes et al., 2013). Regardless by which mechanism mtDNA is replicated, the need for mtRNApol in this process is crucial (for a more detailed review of the interface between transcription and replication in animal mitochondria, we recommend Kasiviswanathan et al., 2012).
mtSSB finalizes our list of proteins to be addressed in this review, possibly with one of the most important roles at the mtDNA replication fork: coordinating interactions of ssDNA, pol γ and Twinkle. It is believed that ssDNA wraps around mtSSB, like the stitches of a baseball, with multiple points of interactions (Oliveira and Kaguni, 2011). In fact, disturbing some of the most conserved residues of the ssDNA-binding domain by alanine substitutions disrupted the ssDNA-binding properties of recombinant mtSSB, as expected (Farr et al., 2004). Remarkably, these mutants were also defective in stimulating the DNA polymerase and exonuclease activities of pol γ. The ability of mtSSB to stimulate in vitro the intrinsic activities of pol y and Twinkle has been shown in individual enzymatic assays (Farr et al., 1999, 2004; Korhonen et al., 2003; Oliveira and Kaguni, 2010, 2011) and in the replisome reconstitution assay (Korhonen et al., 2004). Interestingly, the residues of mtSSB that appear to be important for pol γ and Twinkle stimulation are distinct, indicating that the protein interacts with its partners at the mtDNA replication fork via different mechanisms (Oliveira and Kaguni, 2011). That is somewhat understandable because in the mtSSB-Twinkle interactions, the unwinding of dsDNA by Twinkle releases ssDNA, allowing mtSSB binding; whereas in the mtSSBpol γ interactions, mtSSB is already bound to ssDNA and needs to be displaced from it to allow pol γ access to the template. We can speculate that mtSSB coordinates the functions of these two enzymes, ensuring that the replication fork progresses smoothly during mtDNA duplication. Obviously, further biochemical and physiological data are warranted to test this hypothesis, and to show the possible roles of mtSSB-pol γ interactions in human diseases, as we have proposed previously (Oliveira and Kaguni, 2011).
Concluding Remarks
The goal of the research efforts on mtDNA replication is to understand the basic cellular mechanism(s) that promote(s) duplication of this genome, ultimately providing insight into treatment options for human patients with mtDNA replication-related diseases. However, as research in this area advances, a complex and diverse picture of these mechanisms emerges, giving the impression that treatment is far from being achieved. Nevertheless, it is plausible to speculate that the diversity and complexity of current models for mtDNA replication may reflect the different modes that operate in different tissues/cell types, and represent adaptive processes to ensure appropriate mtDNA copy number and mitochondrial gene expression, and hence ATP production via OXPHOS. Therefore, these findings need to be taken into careful consideration when developing treatments. One may never have thought that describing the replication of a ~16 kb circular genome would apparently be so complicated, involve proteins of strange evolutionary origins, and yet be so important for the health of humans and other animals.
Acknowledgments
We thank Crassos Caio de Oliveira for helping with the figures, and Dr. Mike Gerards for critical reading of the manuscript.
Internet Resources
Received: May 19, 2013; Accepted: July 11, 2013.
Associate Editor: Carlos F.M. Menck
License information: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Bogenhagen D, Gillum AM, Martens PA and Clayton DA (1979) Replication of mouse L-cell mitochondrial DNA. Cold Spring Harb Symp Quant Biol 43:253-262.
- Bogenhagen DF and Clayton DA (2003a) The mitochondrial DNA replication bubble has not burst. Trends Biochem Sci 28:357-360.
- Bogenhagen DF and Clayton DA (2003b) Concluding remarks: The mitochondrial DNA replication bubble has not burst. Trends Biochem Sci 28:404-405.
- Bowmaker M, Yang MY, Yasukawa T, Reyes A, Jacobs HT, Huberman JA and Holt IJ (2003) Mammalian mitochondrial DNA replicates bidirectionally from an initiation zone. J Biol Chem 278:50961-50969.
- Bratic A and Larsson NG (2013) The role of mitochondria in aging. J Clin Invest 123:951-957.
- Brown TA, Cecconi C, Tkachuk AN, Bustamante C and Clayton DA (2005) Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev 19:2466-2476.
- Camara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, et al. (2011) MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab 13:527-539.
- Cheok CF, Wu L, Garcia PL, Janscak P and Hickson ID (2005) The Bloom's syndrome helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res 33:3932-3941.
- Cohen BH and Naviaux RK (2010) The clinical diagnosis of POLG disease and other mitochondrial DNA depletion disorders. Methods 51:364-373.
- Edgar D and Trifunovic A (2009) The mtDNA mutator mouse: Dissecting mitochondrial involvement in aging. Aging 1:1028-1032.
- Euro L, Farnum GA, Palin E, Suomalainen A and Kaguni LS (2011) Clustering of Alpers disease mutations and catalytic defects in biochemical variants reveal new features of molecular mechanism of the human mitochondrial replicase, Pol gamma. Nucleic Acids Res 39:9072-9084.
- Fan L, Sanschagrin PC, Kaguni LS and Kuhn LA (1999) The accessory subunit of mtDNA polymerase shares structural homology with aminoacyl-tRNA synthetases: Implications for a dual role as a primer recognition factor and processivity clamp. Proc Natl Acad Sci USA 96:9527-9532.
- Fan L, Kim S, Farr CL, Schaefer KT, Randolph KM, Tainer JA and Kaguni LS (2006) A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J Mol Biol 358:1229-1243.
- Farge G, Holmlund, T, Khvorostova J, Rofougaran R, Hofer A and Falkenberg M (2008) The N-terminal domain of TWINKLE contributes to single-stranded DNA binding and DNA helicase activities. Nucleic Acids Res 36:393-403.
- Farr CL, Wang Y and Kaguni LS (1999) Functional interactions of mitochondrial DNA polymerase and single-stranded DNA-binding protein. Template-primer DNA binding and initiation and elongation of DNA strand synthesis. J Biol Chem 274:14779-14785.
- Farr CL, Matsushima Y, Lagina AT, Luo N and Kaguni LS (2004) Physiological and biochemical defects in functional interactions of mitochondrial DNA polymerase and DNA-binding mutants of single-stranded DNA-binding protein. J Biol Chem 279:17047-17053.
- Fuste JM, Wanrooij S, Jemt E, Granycome CE, Cluett TJ, Shi Y, Atanassova N, Holt IJ, Gustafsson CM and Falkenberg M (2010) Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol Cell 37:67-78.
- Holt IJ (2009) Mitochondrial DNA replication and repair: All a flap. Trends Biochem Sci 34:358-365.
- Holt IJ, Lorimer HE and Jacobs HT (2000) Coupled leading-and lagging-strand synthesis of mammalian mitochondrial DNA. Cell 100:515-524.
- Jemt E, Farge G, Backstrom S, Holmlund T, Gustafsson CM and Falkenberg M (2011) The mitochondrial DNA helicase TWINKLE can assemble on a closed circular template and support initiation of DNA synthesis. Nucleic Acids Res 39:9238-9249.
- Joers P and Jacobs HT (2013) Analysis of replication intermediates indicates that Drosophila melanogaster mitochondrial DNA replicates by a strand-coupled theta mechanism. PLoS One 8:e53249.
- Kang D, Miyako K, Kai Y, Irie T and Takeshige K (1997) In vivo determination of replication origins of human mitochondrial DNA by ligation-mediated polymerase chain reaction. J Biol Chem 272:15275-15279.
- Kasiviswanathan R, Collins TR and Copeland WC (2012) The interface of transcription and DNA replication in the mitochondria. Biochim Biophys Acta 1819:970-978.
- Korhonen JA, Gaspari M and Falkenberg M (2003) TWINKLE Has 5'->3'DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J Biol Chem 278:48627-48632.
- Korhonen JA, Pham XH, Pellegrini M and Falkenberg M (2004) Reconstitution of a minimal mtDNA replisome in vitro. EMBO J 23:2423-2429.
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481-484.
- Martin-Negrier ML, Sole G, Jardel C, Vital C, Ferrer X and Vital A (2011) TWINKLE gene mutation: Report of a French family with an autosomal dominant progressive external ophthalmoplegia and literature review. Eur J Neurol 18:436-441.
- Matsushima Y and Kaguni LS (2007) Differential phenotypes of active site and human autosomal dominant progressive external ophthalmoplegia mutations in Drosophila mitochondrial DNA helicase expressed in Schneider cells. J Biol Chem 282:9436-9444.
- Matsushima Y and Kaguni LS (2009) Functional importance of the conserved N-terminal domain of the mitochondrial replicative DNA helicase. Biochim Biophys Acta 1787:290-295.
- Matsushima Y, Farr CL, Fan L and Kaguni LS (2008) Physiological and biochemical defects in carboxyl-terminal mutants of mitochondrial DNA helicase. J Biol Chem 283:2396423971.
- Milenkovic D, Matic S, Kuhl I, Ruzzenente B, Freyer C, Jemt E, Park CB, Falkenberg M and Larsson NG (2013) TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum Mol Genet 22:1983-1993.
- Nass MM (1980a) Analysis of the two heavy and light strand origins and the direction of replication of mitochondrial DNA within a detailed physical map of this genome in transformed hamster cells. J Mol Biol 140:231-256.
- Nass MM (1980b) Pulse-label analysis and mapping of the two terminal regions of asynchronous complementary strand replication of mitochondrial DNA in transformed hamster cells. J Mol Biol 140:257-281.
- Nunnari J and Suomalainen A (2012) Mitochondria: In sickness and in health. Cell 148:1145-1159.
- Oliveira MT and Kaguni LS (2010) Functional roles of the N-and C-terminal regions of the human mitochondrial singlestranded DNA-binding protein. PLoS One 5:e15379.
- Oliveira MT and Kaguni LS (2011) Reduced stimulation of recombinant DNA polymerase gamma and mitochondrial DNA (mtDNA) helicase by variants of mitochondrial single-stranded DNA-binding protein (mtSSB) correlates with defects in mtDNA replication in animal cells. J Biol Chem 286:40649-40658.
- Oliveira MT, Garesse R and Kaguni LS (2010) Animal models of mitochondrial DNA transactions in disease and ageing. Exp Gerontol 45:489-502.
- Pohjoismaki JL, Goffart S, Tyynismaa H, Willcox S, Ide T, Kang D, Suomalainen A, Karhunen PJ, Griffith JD, Holt IJ, et al. (2009) Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J Biol Chem 284:2144621457.
- Pohjoismaki JL, Holmes JB, Wood SR, Yang MY, Yasukawa T, Reyes A, Bailey LJ, Cluett TJ, Goffart S, Willcox S, et al. (2010) Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. J Mol Biol 397:1144-1155.
- Reyes A, Yang MY, Bowmaker M and Holt IJ (2005) Bidirectional replication initiates at sites throughout the mitochondrial genome of birds. J Biol Chem 280:3242-3250.
- Reyes A, Kazak L, Wood SR, Yasukawa T, Jacobs HT and Holt IJ (2013) Mitochondrial DNA replication proceeds via a 'bootlace' mechanism involving the incorporation of processed transcripts. Nucleic Acids Res 41:5837-5850.
- Robberson DL, Kasamatsu H and Vinograd J (1972) Replication of mitochondrial DNA. Circular replicative intermediates in mouse L cells. Proc Natl Acad Sci USA 69:737-741.
- Scheffler IE (2008) Mitochondria. 2nd edition. J. Wiley and Sons, Inc., Hoboken, 462 pp.
- Sen D, Nandakumar D, Tang GQ and Patel SS (2012) Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J Biol Chem 287:14545-14556.
- Shutt TE and Gray MW (2006a) Bacteriophage origins of mitochondrial replication and transcription proteins. Trends Genet 22:90-95.
- Shutt TE and Gray MW (2006b) Twinkle, the mitochondrial replicative DNA helicase, is widespread in the eukaryotic radiation and may also be the mitochondrial DNA primase in most eukaryotes. J Mol Evol 62:588-599.
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, et al. (2001) Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet 28:223-231.
- Stumpf JD and Copeland WC (2011) Mitochondrial DNA replication and disease: Insights from DNA polymerase gamma mutations. Cell Mol Life Sci 68:219-233.
- Stumpf JD, Saneto RP and Copeland WC (2013) Clinical and molecular features of POLG-related mitochondrial disease. Cold Spring Harb Perspect Biol 5:a011395.
- Tapper DP and Clayton DA (1981) Mechanism of replication of human mitochondrial DNA. Localization of the 5' ends of nascent daughter strands. J Biol Chem 256:5109-5115.
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, et al. (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417-423.
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA and Loeb LA (2007) Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet 39:540-543.
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA and Loeb LA (2008) DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet 40:392-394.
- Wanrooij S, Goffart S, Pohjoismaki JL, Yasukawa T and Spelbrink JN (2007) Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res 35:3238-3251.
- Wanrooij S, Fuste JM, Farge G, Shi Y, Gustafsson CM and Falkenberg M (2008) Human mitochondrial RNA polymerase primes lagging-strand DNA synthesis in vitro Proc Natl Acad Sci USA 105:11122-11127.
- Yang MY, Bowmaker M, Reyes A, Vergani L, Angeli P, Gringeri E, Jacobs HT and Holt IJ (2002) Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell 111:495-505.
- Yasukawa T, Reyes A, Cluett TJ, Yang MY, Bowmaker M, Jacobs HT and Holt IJ (2006) Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J 25:5358-5371.
- Ziebarth TD, Gonzalez-Soltero R, Makowska-Grzyska MM, Nunez-Ramirez R, Carazo JM and Kaguni LS (2010) Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J Biol Chem 285:1463914647.
- Human DNA Polymerase Gamma Mutation Database, http://tools.niehs.nih.gov/polg (April, 2013).
» link - RCSB Protein Data Bank, http://www.rcsb.org/pdb/home/home.do (April, 2013).
» link
Send correspondence to
Publication Dates
-
Publication in this collection
23 Aug 2013 -
Date of issue
2013
History
-
Received
19 May 2013 -
Accepted
11 July 2013