Abstract
The melting and solidification behavior of Bi2Sr2Ca2Cu3 O10 (Bi-2223) precursors has been studied. Nominal compositions corresponding to excess of liquid, Ca2CuO3 and CuO have been investigated. Each sample was made by packing a precursor powder into a silver crucible, in order to approximately simulate the situation found in 2223 silver-sheathed tapes. The samples were partially melted and then slow-cooled, being quenched from different temperatures and analyzed through X-ray diffraction (XRD) and scanning electron microscopy (SEM/EDS). The precursors decomposed peritectically during melting, forming liquid and solid phases. Very long plates with compositions falling in the vicinity of the 2223 primary phase field formed upon slow-cooling. The 2223 phase may have been formed and the results suggest that long grains of this phase might be obtained by melting and crystallization if the exact peritectic region and the optimum processing conditions are found.
high temperature superconductors; Bi-2223; melting and crystallization
REGULAR ARTICLES
Phase relations study on the melting and crystallization regions of the Bi-2223 high temperature superconductor
Alexander PolasekI, * * e-mail: polasek@cepel.br ; Peter MajewskiII; Eduardo Torres SerraI; Fernando RizzoIII; Fritz AldingerIV
ICEPEL-Electric Power Research Center, C.P. 68007, 21944-970 Rio de Janeiro - RJ, Brazil
IIUniversity of South Australia, Ian Wark Research Institute, Mawson Lakes South Australia 5095, Adelaide, Australia
IIIPUC-Rio, Pontifical Catholic University, Department of Materials Science and Metallurgy, C.P. 38008, 22453-900, Rio de Janeiro - RJ, Brazil
IVMax-Planck-Institut für Metallforschung, Pulvermetallurgisches Laboratorium, 70569 Stuttgart, Germany
ABSTRACT
The melting and solidification behavior of Bi2Sr2Ca2Cu3 O10 (Bi-2223) precursors has been studied. Nominal compositions corresponding to excess of liquid, Ca2CuO3 and CuO have been investigated. Each sample was made by packing a precursor powder into a silver crucible, in order to approximately simulate the situation found in 2223 silver-sheathed tapes. The samples were partially melted and then slow-cooled, being quenched from different temperatures and analyzed through X-ray diffraction (XRD) and scanning electron microscopy (SEM/EDS). The precursors decomposed peritectically during melting, forming liquid and solid phases. Very long plates with compositions falling in the vicinity of the 2223 primary phase field formed upon slow-cooling. The 2223 phase may have been formed and the results suggest that long grains of this phase might be obtained by melting and crystallization if the exact peritectic region and the optimum processing conditions are found.
Keywords: high temperature superconductors, Bi-2223, melting and crystallization
1. Introduction
The advent of superconductivity above the liquid nitrogen temperature (77 K) has greatly increased the potential for applications and commercialization of superconductors. However, all known "liquid nitrogen superconductors" are brittle copper-based oxides, with a highly anisotropic current transport1. Among these materials, the oxide Bi2Sr2Ca2Cu3 O10+x (Bi-2223) has been the most suitable for manufacturing long wires and cables2,3. Its high critical transition temperature (Tc = 110 K), combined with its ability to attain high texture degrees in the appropriate current transport direction, allow high critical current densities at the liquid nitrogen temperature (> 104A/cm2).
The Bi-2223 phase belongs to the Bi-Sr-Ca-Cu-O system, which also includes the Bi2Sr2CuO6 (Bi-2201, Tc < 20 K) and the Bi2Sr2CaCu2O8 (Bi-2212, Tc < 96 K) phases4-6. In fact, they are solid solutions, forming with compositions deviating from the stoichiometric ones. The superconducting Bi2Sr2CuO6, also known as Raveau-phase, can be better represented as Bi11Sr9CaxCu5 Ox (119 × 5) and is frequently confused with the non-superconducting 2201 phase, which has a different structure7. Considering the close proximity of these phases and for the sake of simplicity, both will be referred as 2201 througout the present work. The 2201, 2212 and 2223 grains have a plate-like morphology with high aspect ratios, enabling the appropriate orientation to be attained2,3,5.
Wires of 2223 are made by inserting a pre-reacted precursor powder into a silver tube that is further mechanically deformed into a flat tape; this composite tape is then subjected to a long heat treatment process, in order to convert the silver sheathed powder into the 2223 phase3. Intermediate cold-rolling steps are performed to improve the reaction and the grain alignment. For more than one decade, many efforts have been made to improve the critical current densities of these composite tapes and prototypes of equipments and devices have been successfully demonstrated worldwide2-4,8.
In spite of the remarkable progresses achieved, however, the processing costs remain still quite high for large-scale commercialization, mainly because of the silver sheath8. This metal is employed due to its suitable equilibrium properties at the processing temperatures, high ductility and oxygen permeability. In addition, silver is thought to enhance the 2223 phase formation and grain alignment9. The large-scale application of 2223 tapes in the electric power industry depends mainly on the achievement of high critical current densities, in order to improve the cost/performance of such tapes. It has been shown that the local current transport of the tapes is extremely inhomogeneous, indicating that their microstructure is far from perfect10. The improvement of the grain connectivity and alignment is essential for increasing the critical current density. Therefore, one can expect a significant enhancement in the transport properties of the tapes by optimizing the manufacturing procedures.
Conventionally, the 2223 phase is synthesized by long term sintering at temperatures close to the melting4-6. Nevertheless, the full sintered ceramic normally presents a high porosity and many cracks, decreasing the current transport capacity11,12. Another hampering factor is the complex multiphase equilibrium involved, leading to the formation of secondary-phases that disturb the alignment and coupling of the superconducting plates13,14.
Alternatively, a melt-processing route may lead to higher bulk densities, better texture and longer superconducting plates. The melt-processing consists of partially melting a precursor powder, with subsequent slow-cooling and further annealing. The 2212 phase is routinely melt-processed, being peritectically decomposed into liquid and solid phases, and then recrystallized through the inverse peritectic reaction by further slow cooling15,16. Attempts to grow 2223 from the melt have already been done, but its narrow stability range and sluggish formation kinetics prevent its crystallization from the melt17-20. Besides, there is a lack of knowledge on the concentration region where 2223 is in equilibrium with the liquid, but some works indicated the feasibility of the 2223 crystallization from the melt20-25. Giannini et al.25 have recently observed the 2223 reformation directly from a reaction involving melt and solid phases, though in samples where the 2223 phase was not fully melted.
The present work has the aim of better understanding the phase relationships involved in the 2223 melting and crystallization regions, as well as to investigate the feasibility of 2223 crystallization from temperatures above the liquidus line. Pre-reacted precursor powders have been packed into silver crucibles and then melted, slowly cooled and quenched. Silver is employed to approximately simulate the situation found in 2223 silver-sheathed tapes. Nominal compositions corresponding to excess of liquid, Ca2CuO3 (2:1) and CuO have been investigated. The 2:1 and CuO phases play important roles, acting as Ca and Cu sources to convert 2212 into 2223 by sintering4-6. Although the 2223 formation by sintering is considerably improved by lead doping4, the high volatility of this element can be a critical limiting factor for melt-processing. Therefore, lead has not been employed in the present study. All melting-crystallization experiments have been performed under 0.08 atm O2, the oxygen partial pressure normally employed in tapes processing, because it maximizes the 2223 stability range2, 3. Our results indicate that 2223 might form during the slow cooling, and suggest that long grains of this phase may be obtained by melt processing if the 2223 peritectic equilibrium can be well defined.
2. Experimental
The reagents Bi2O3 (ALDRICH 99.9%), SrCO3 (ALDRICH 98 + %), CaCO3 (ALDRICH 99 + %) and CuO (ALDRICH 99 %) were dry mixed and homogenized in an agate mortar with pestle, in the nominal compositions Bi2Sr1.2Ca2.4Cu3O x (A), BiSrCa2.5Cu3Ox (B) and Bi3.37Sr2.1Ca2.17Cu3O x (C). These compositions lie within phase regions including excess of Ca2CuO3 (2:1 phase), CuO and melt. The starting mixtures were calcined for 48 h in air, between 750 ºC and 800 ºC, with intermediate grinding steps. The full calcined powders were isostatically cold pressed and sintered for 180 h in air, at 820-840 ºC, with intermediate regrinding and repressing steps. All calcinations and sinterizations were performed in box furnaces with alumina boats. Monitoring powders composition by Ion-Coupled-Plasma (ICP-OES) revealed no appreciable variation in the starting cation proportions throughout precursors preparation.
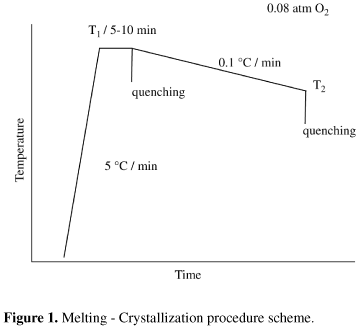
For the melting-crystallization treatments, the full-processed precursors were packed into silver crucibles. The silver crucibles consisted of high-purity silver (99.99%) cylinders measuring 8.0 mm (outer length) × 6.0 mm (outer diameter) and 3.0 mm (inner diameter) × 3.0 mm (inner length). In order to avoid bismuth and oxygen volatilization, the top of each crucible was closed with a silver disk measuring 6.0 mm (diameter) × 2.0 mm (thickness) before the melting-crystallization heat treatments. Yttrium disks were placed on the bottom of the alumina boat for protecting the crucible from reactions with the melt. The samples were heated for 5-10 min at the maximum temperature, subsequently slow-cooled and quenched, as shown in Fig. 1. The quenching was performed by taking the silver crucible out of the furnace and placing it on a thick copper plate at room temperature. The melting-crystallization treatments were carried out in a tubular furnace, under 0.08 atm O2 balanced with N2. All temperatures were measured within +/- 5 ºC, with cromel-alumel thermocouples calibrated against the silver melting point (961.8 ºC).
In order to study the microstructure of the ceramic core, the polished cross-section of the quenched samples was characterized by use of scanning electron microscopy with X-ray energy dispersive analysis (SEM / EDS - Cambridge Instruments S 200 and Zeiss Gemini). Phase identification was performed by means of X-ray diffraction analysis with l CuKa radiation (XRD - Siemens D-5000 and Diano XRD 8545). Excepting the precursors, the XRD samples were obtained by scrapping and grinding the ceramic core of the quenched samples. In order to evaluate the transition temperatures, differential thermal analysis (DTA) was carried out on samples consisting of precursor powders (0.01 g) packed into silver crucibles measuring 6.0 mm (outer diameter) × 1.5 mm (thickness) and 2.0 mm (inner diameter) × 1.0 mm (inner length).
3. Results
3.1. Precursors
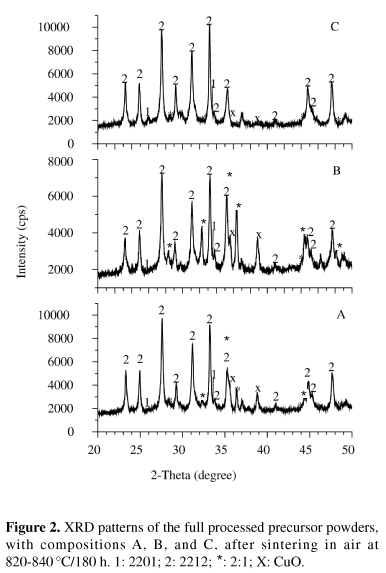
Almost the same phase assemblage has been obtained by treating precursors with very different overall compositions in a relatively wide temperature range. As shown in Fig. 2, the 2212 phase is preponderant in all sintered precursors, together with significant amounts of (Ca,Sr)2CuO3 (2:1), CuO and traces of 2201. Such phase assemblage was expected to take place at the sintering temperatures employed. In good agreement with the cation stoichiometries employed herein, the highest fractions of 2:1 and CuO have been found in the precursor B, while the precursor C presents the lowest fractions of those phases.
3.2. Thermal Analysis
Figure 3 shows differential-thermal-analysis (DTA) scans, in the range of 800-895 ºC, taken from silver-packed precursors. No relevant events were observed at lower temperatures24. After each DTA run, the sample seemed to have melted. Since the 2:1 and CuO phases melt only above 950 ºC, the endothermic peak should correspond to a partial melting of the precursors. The intensity of this endothermic peak varies with the composition, scaling with the liquid amount, since the initial powder amount was almost the same for each sample. In fact, for all melting treatments carried out, the aspect of the quenched samples indicated that the samples C formed the highest liquid amounts, whereas the samples B formed the lowest ones.
The melting temperatures have been determined through the intersection of the baseline with the tangents of the main endothermic peaks. The melting onset situates at 840-845 ºC and the melting outset is located at 865-870 ºC, showing that the melt occurs within a range instead of a fixed point, as expected for a peritectic decomposition.
Two main exothermic peaks seem to have arisen during slow cooling, at 870-880 ºC and at 825-835 ºC, possibly indicating phase formation. It can hardly be noticed for composition B, for which it may have appeared below 830 ºC. It is remarkable that the first slow-cooling peak has always risen above the melting range, whereas the second peak has always risen below that range. Nevertheless, it is important to recall that the samples were heated at 5 ºC/min and slow-cooled at 0.5 ºC/min. The heating and cooling rates can affect the position and intensity of the peaks. In general, a peak tends to become more intense and sharper for high rates, which can explain the small crystallization peaks found here. The slow-cooling rate was used to simulate the situation of the melting-crystallization treatments (Fig. 1). Furthermore, events that can take place at low rates may not occur at higher rates, due to kinetical reasons.
3.3. Melting-Crystallization at 900-815 ºC, in 0.08 atm O2
The precursors A, B and C were packed into silver crucibles for the melting-crystallization treatments, in 0.08 atm O2 balanced with N2. Initially, the samples were heated at 875 ºC for 5 min and quenched, while others were heated at 900 ºC for 5 min and also quenched (Fig. 4). Afterwards, silver-packed precursors were heated at 900 ºC for 5 min, slow cooled (0.1 ºC / min) to 850 ºC or to 815 ºC, and then quenched (Fig. 5).
In contrast with the precursors, no 2212 has been detected after these treatments. The 2201 is the main phase in all quenched samples, with the exception of the samples B, whose 2:1 amounts are comparable to those of 2201. The qualitative comparison among the different compositions reveals a similar feature to that found in the precursors (Fig. 2): the highest 2:1 and CuO fractions are found in the samples B, while the samples C present the lowest amounts of these phases. Silver arises from the sample preparation by scrapping the ceramic core from the silver crucible.
Some features can be noticed by accompanying the phase evolution as illustrated by Figs. 2, 4 and 5. In the first place, regardless of the precursor overall composition, the 2212 decomposed and it did not recrystallize through slow cooling, even in samples cooled to 815 ºC, under 0.08 atm O2, that is, within the 2212 stability range. Second, calcium-rich 2201 and 2:1 seems to be the main products from the 2212 peritectic melting and subsequent quenching. The 2:1 amounts clearly increased in comparison with the precursors but the CuO contents seem to have only slightly decreased during melting. Beside of the mentioned phases, small CaO peaks arose in the sample C heated up to 900 ºC. By cooling down to 850 ºC, the 2:1 fractions seem to have remained almost constant, decreasing in the samples A and B cooled down to 815 ºC. The CuO contents remained apparently constant by cooling down to 850 ºC, but practically vanished at 815 ºC.
The SEM images resulting from the same treatments described above are shown in Figs. 6 and 7. The EDS results are consistent with the previous XRD observations. The light-gray matrix and the white plates exhibited Bi1.72.5(Sr,Ca)1.2-1.7CuOx compositions, falling within the 2201 calcium-rich region. The 2201 phase is known to precipitate very fast from the melt21, and the matrix may thus consist of a mixture of amorphous phase with crystallized 2201 plates or just the latter. However, the analysis carried out in the present work could not clarify this point. In addition, a second matrix composition lying within Bi2.53.75Sr11.18Ca1-1.63 Cu2.59-2.9Ox has been found in darker regions of the matrix of the samples with composition C.
As in the previous XRD patterns, the main secondary phases found by SEM/EDS are 2:1, with composition Ca2-xSrxCuO2 (x = 0.05 0.2), and CuO. Large CaO precipitates have been also identified in the sample C quenched from 900 ºC, though just a low amount of this phase can be observed in the corresponding XRD pattern (Fig. 4). The dendrites found in the sample C slow-cooled to 850 ºC, and in the sample A slow-cooled to 815 ºC, have been associated with the CuO phase. The identification of such fine dendrites is, however, limited by the EDS resolution (12 µm).
High fractions of large secondary-phases (up to 20 µm) can be seen in the samples A and B heated at 875 ºC and 900 ºC. For the composition B, in particular, this amount increased substantially from the samples heated at 875 ºC to the samples heated at 900 ºC. Most of these secondary-phase islands have been identified as the 2:1 phase and some as CuO. These particles enlarged through slow cooling to 850 ºC and most of them were consumed in the sample A slowly cooled to 815 ºC, but many large grains remained in the sample B slow-cooled to the same temperature (Fig. 7). Long light-gray plates and darker regions can be observed for the sample A slowly cooled to 815 ºC. In opposition to the compositions A and B, the samples C exhibited smaller quantities of secondary-phases, in spite of some large precipitates. Initially, dispersed 2:1 precipitates rose at 875 ºC, in a Ca-rich 2201 matrix. These precipitates were almost totally consumed, whereas very large CaO precipitates emerged at 900 ºC. Long Ca-rich 2201 plates embedded in darker regions of the matrix with compositions Bi2.53.75Sr11.18Ca1-1.63 Cu2.59-2.9Ox, together with CuO dendrites, can be viewed in the micrograph corresponding to the slow cooling to 850 ºC. The small dendrites remained upon slow cooling to 815 ºC, where some large 2:1 precipitates were also present.
3.4. Melting-Crystallization at 855-825 ºC, in 0.08 atm O2
In another set of experiments, silver-packed precursors were heated up to 855 ºC and slow cooled down to 825 ºC, in 0.08 atm O2, at the same rate applied before (0.1 ºC/min). This new temperature range has been chosen with basis on the previous results, i.e., a maximum temperature of 900 ºC appeared to be excessively high, preventing not only the formation of 2223 but even of 2212, and generating high-fractions of coarse secondary-phase grains. The final slow-cooling temperature of 825 ºC lies within the 2223 stability range. The composition B was discharged due to excessive segregation of 2:1 and CuO.
Similarly to the previous melting treatments, Ca-rich 2201, 2:1 and CuO have been found. Again, the 2:1 fractions clearly increased in comparison to the precursors, indicating that this phase precipitated from the 2212 decomposition. Nevertheless, less segregation seems to have occurred. Some 2212 was present in the samples quenched from 855 ºC (Fig. 8), without a slow cooling step. This indicates that either the 2212 present in the precursor had not completely melted or some 2212 may have precipitated during quenching. After slow cooling, the 2201 remained as the main phase. The XRD pattern of the slow-cooled sample A shows no clear indications of 2212 or 2223 formation, whereas the slow-cooled sample C gave rise to significant 2212 fractions and possibly also 2223.
The corresponding SEM images are shown in Fig. 9. As had been observed after the treatments at higher temperatures, both the molten and the slow-cooled specimens with composition A showed coarse 2:1 and CuO islands embedded in a Bi1.7-2.5(Sr,Ca)1.2-1.7CuOx (Ca-rich 2201) matrix. The samples with composition C exhibited much less and smaller 2:1 and CuO islands, within a similar matrix. The samples quenched from the melt exhibit an apparently heterogeneous matrix, specially for composition C. An image of the same specimen, with a higher magnification, reveals that those regions roughly have a plate-like shape, with not well defined contours (Fig. 10). Their shape and small size suggest that a partial melting or a partial crystallization took place. The EDS analysis of such "plates" gave Bi2528Sr17-21Ca19-23 Cu29-33Ox compositions, falling in the vicinity of the 2223 primary phase-field. Such compositions may indicate the presence of either 2223 or a 2212 phase with a low Sr:Ca ratio, as well as a mixture of both phases. However, the corresponding XRD pattern indicates only the presence of 2212. There is still an interesting feature in Fig. 10, namely the presence of narrow white regions in the vicinity of the "plates". The EDS resolution cannot distinguish these regions from the matrix, but its tonality suggests a Ca-rich 2201 composition with a lower Ca content than that of the matrix.
The plates found in the melted samples grew during slow cooling (Fig. 9), maintaining similar compositions. This time, however, the XRD result of composition C has also pointed 2223. Interestingly, the XRD pattern of composition A has pointed neither 2223 nor 2212. From the tonality and compositions of the plates, and the identification of 2212 and possibly also 2223 for the C composition, it is very unlikely that the plates found in the sample A belong to the Ca-rich 2201. This leads to the conclusion that the 2212 and 2223 peaks might be overlapped by the broad and strong 2201 peaks.
4. Discussion
4.1. Thermal analysis
The endothermic peak in the DTA heating curves (Fig. 3), situated at 840-870 ºC, should correspond to the peritectic melting of 2212, since it was the preponderant phase in the precursors and is known to decompose below 900 ºC in air, whereas the 2:1 and CuO secondary-phases melt only above 950 ºC in air. The 2212 phase melts incongruently within a temperature range depending on the composition. For Sr:Ca ~ 2, the 2212 phase is reported to decompose at 890900 ºC in air26. With decreasing the Sr:Ca ratio that temperature lowers down to about 830 ºC. The low DTA onset temperatures indicate a low Sr:Ca ratio in the 2212 present in the precursors. The liquid fraction has been found to vary considerably with the precursor composition, revealing a good agreement with the phase diagram. Composition B, lying within a region very rich in 2:1 and CuO, has given the shallowest endothermic peak, while composition C, falling in a region rich in liquid, has leaded to the deepest endothermic peak. Composition A, which corresponds to a somewhat intermediary situation, has indeed produced an endothermic peak with intensity lying in between those of B and C.
However, the composition effect seems not to account for the low outset temperatures. The temperature for the complete decomposition of 2212 lies above 890 ºC, regardless of the Sr:Ca ratio. As silver is a further factor that can lower the 2212 decomposition temperatures, the use of silver crucibles may have also affected the melting. The temperature for complete decomposition can decrease from 895 ºC down to 865 ºC, with increasing Ag concentrations up to about 40 mol %27. Furthermore, silver may have also played an important role in the onset temperature, since it can also reduce the 2212 Tsolidus.
Two main exothermic peaks appeared during slow-cooling, being possibly related to the crystallization of the 2201 and 2212 phases. In a melting study with high-purity 2212 samples, Neves et al.28 showed that the 2201 crystallization precedes the 2212 reformation upon cooling, generating a similar situation in their DTA cooling curves.
Thermal Gravimetric Analysis (TGA) revealed low average mass variation upon melting and mass gain during the slow-cooling step, for samples with the same compositions employed in the present work24. Normally, the mass is expected to decrease by melting, due to losses of volatile elements such as bismuth and oxygen. However, the silver disks used to close the crucibles may have avoided such losses during melting24. Besides, silver can retain oxygen in the liquid phase29. Since the liquid dissolves a certain amount of silver, the use of silver crucibles can have provided this element, avoiding mass losses by melting. Such effect can be advantageous for melting-process, because the oxygen content is critical for the formation and stability of the superconducting phases. The 2212 phase is known to absorb oxygen from the atmosphere by cooling, which possibly explains the mass increasing during slow cooling24.
4.2. Melting-Crystallization
Initially, the precursors sintered at 820-840 ºC in air consisted mainly of the 2212, Ca2CuO3 (2:1 phase), CuO phases and traces of 2201, a phase assemblage expected to take place at temperatures below the 2223 equilibrium range (850-890 ºC in air). By melting and quenching, this phase assemblage evolved to large 2:1 and CuO islands within a Ca-rich 2201 matrix. This agrees well with previous investigations on the melting-crystallization relations in the 2223 phase field19,20,25,31-33. Although 2223 was not present in the precursors, their overall compositions pertain to regions situated around the 2223 single-phase field. Figure 11 illustrates the nominal compositions of the precursors projected on the phase diagram representing an isothermal section of the Bi2O3-SrO-CaO-CuO system at 850 ºC in air34,35. This section is of primary importance, since it passes through the 2223 single-phase region. It is important to notice, however, that this diagram corresponds to a temperature well below the 2212 and 2223 liquidus lines. Comparisons with the present work should also take into account that the melting temperatures are considerably reduced in 0.08 atm O2, as well as in the presence of silver. Composition B lies within a Ca- and Cu- rich region, i.e., with high fractions of Ca-Cu-O phases; in the other extreme, composition C falls within a Bi-rich region, that is, closer to the liquid field, while composition A is in an intermediary situation. Indeed, the samples with compositions A and C exhibited strong indications of melting. On the contrary, much less liquid seemed to have formed in the samples with composition B, though it also melted as well. Strobel et al.36 found similar features in molten samples, relating this to the fact that the bismuth rich-side of the phase diagram contains higher fractions of superconducting BSCCO phases than the bismuth poor-side, which includes more Ca-Cu-O phases. Since these phases remain solid in the range of temperatures employed in this work, less liquid was produced in samples rich in Ca and Cu. All compositions fall within multiphase fields, where 2223 coexists with some or all of the following phases: 2212, 2:1, 14:24, CuO and liquid. The fact that 14:24 has not been found in our samples is probably associated to its decomposition temperature being lowered in 0.08 atm O2, as well as to its limited stability range29. This phase has been found as an equilibrium product of the 2223 decomposition and reformation, but in little amounts25,32. Besides, the 2:1 phase has been shown to be the most important secondary-phase involved in the melting-crystallization relations of 2223 unleaded samples20,23-25,32,33, which agrees very well with the results of the present work.
The main phase present in the precursors, the 2212, decomposes into melt and 2:1, as observed through high-temperature microscopy in samples with 2223 nominal composition33. In samples with compositions closer to the 2212 concentration region, the decomposition reaction of this phase generates also bismuthates15,16,29, which were not found here. Therefore, the increase of the 2:1 fractions through melting, with the formation of large precipitates of this phase, can be explained by the 2212 decomposition within the 2223 compositional range. According to the diagram from Fig. 11, the effective stoichiometry of the 2212 phase in equilibrium with 2223 is Bi2Sr1.5Ca1.5Cu2O x. In a phase-diagram study on the subsolidus region, Wong-Ng et al.37 have also found such 2212 type forming adjacent to the 2223 primary crystallization field. The 2212 phase with a Sr:Ca ~ 1 initiates its peritectic melting at relatively low temperatures, whereas specimens with a composition close to Bi2Sr2CaCu2O8 only begins to decompose at temperatures above 880 ºC in air26, which fits with our DTA results indicating melting onset at 840-845 ºC in air. The 2212 and 2223 solidus temperatures decrease with increasing the Ca content, favoring thus the 2223 formation, as long as it has been proved that, in the case of the conventional sintering process, this phase is more efficiently formed above its partial melting line34,35.
The influence of the maximum heating temperature can be clearly observed. The 2:1 amounts increased with the heating temperature, reaching a maximum at 875 ºC for compositions A and C, and at 900 ºC for composition B, for which the 2:1 fraction practically surpassed the 2201 content. Buhl et al.15 found that the maximum temperature and the holding time at this temperature play central roles for melt-processing the 2212 phase. Control of these parameters is essential for avoiding too much segregation and also bismuth and oxygen volatilization. With increasing the temperature and/or the holding time at the maximum temperature, more Ca and Cu can segregate from the melt forming large 2:1 and CuO precipitates. Moreover, too high temperatures and long permanence times may induce bismuth and oxygen volatilization, specially for low O2 partial pressures24. It is possible that the low oxygen partial pressure induced oxygen loss upon melting. Lomello-Tafin38 showed that low oxygen partial pressures induce more weight-loss. In addition, large CaO precipitates formed by melting the precursor C at 900 ºC in 0.08 atm O2. This is attributed to the decomposition of Ca2CuO3 (2:1) into CaO and CuO, taking place above 950 ºC in air31-33. Besides, the silver present in the crucibles can reduce those temperatures by about 10-20 ºC31,32. As no CaO has been observed for the other compositions, the formation of this phase at about 900 ºC can thus be attributed to the combined effects of the oxygen partial pressure, the presence of silver and the composition of the precursor. Such 2:1 decomposition, as well as the excessive 2:1 precipitation in the specimen with composition B heated to 900 ºC indicate that this temperature is too high for melt-processing.
In fact, it is quite interesting that even 2212 has not been formed in the samples slowly cooled from 900 ºC to 815 ºC, since this latter temperature situates well within the 2212 stability range. This finding is more surprising if one considers the very slow cooling rate employed (0.1 ºC/min), as this phase can easily grow from the perictetic melt at even higher cooling rates33. Since heating up to 855 ºC is sufficient to decompose practically all the 2212 of the precursors, and considering the discussion above, it may be concluded that the maximum processing temperature should lie slightly above the liquidus line. For melt-processing the 2212 phase, it has been shown that the maximum temperature should lie at 5-8 ºC above the solidus line15. Yet, the presence of large 2:1 and CuO islands may have also prevented the 2212 and 2223 formation in those samples, due to a lack of contact area between the precipitates and the liquid, as well as to difficulties in dissolving such large precipitates to provide sufficient Sr and Ca to a Bi-rich melt, in the amount necessary to form 2212 and 2223. Another hampering factor is the very fast crystallization of 2201, even during quenching21. It was shown that the fraction of 2201 lowers, while that of 2212 increases as the cooling rate decreases, which can be related to the deficiency of Ca and Cu, and the enrichment of Bi in the liquid phase at the growth fronts of the 2212 and 2223 phases39,40. The microstructure of the sample C quenched from 855 ºC (Fig. 10) suggests 2212 platelets growing within a Bi-rich liquid by absorbing Ca and Cu from the adjacent matrix. It can be argued, however, that such microstructure has only resulted from a residual not fully melted 2212. In order to elucidate this point, high temperature analysis is required.
The exact nature of the liquid in equilibrium with the 2223 phase remains to be elucidated, with some works pointing to a nearly Ca-rich 2201 melt and others suggesting a composition closer to that of the 2212 phase. The answer to this question is critical for understanding the results of the present work and for providing experimental routes that succeed in obtaining high 2223 fractions by melt-processing. Schulze et al.34 pointed out that the melt should be rich in bismuth, having approximately a Bi2(Sr,Ca)1.5CuOx (Ca-rich 2201) composition, since this phase melts congruently and crystallizes directly from the liquid, differently from the 2212 and 2223 phases. According to Majewski27, with increasing temperatures from below to above the melting of 2212 and 2223, the liquid first becomes more Ca-, Cu- and Bi-rich, with concentration of Sr increasing later; at about 850 ºC in air the liquid phase ranges from the Bi-rich concentration region to a composition around Bi2SrCaCuOx. The composition of the melt in equilibrium with 2212 and 2223 was determined to be about Bi2Sr1-1.3Ca0.4-1CuOx and Bi2Sr0.8Ca0.9CuOx, respectively27,34,35. However, other studies revealed a melt with compositions closer to that of the 2212 phase31,32. Park et al.32 found a Bi26.8Sr22.6Ca12.4 Cu31.1Ox melt forming at 860-870 ºC in air, in precursors mixed with silver. Styve et al.31 observed similar compositions.
It was reported that the 2201 and 2212 phases are not equilibrium products from the melting, but both may coprecipitate during cooling25,40. This may lead to the conclusion that the real melt composition falls between the "2212" and "2201" stoichiometries, as already pointed out by Liu et al.40. Although all quenched samples of the present work showed a Ca-rich 2201 matrix, regions with compositions Bi2.5-3.75Sr1-1.18Ca1-1.63 Cu2.59-2.9Ox, i.e., located between the Ca-rich 2201 and the 2212 stoichiometries, have also been found in samples with composition C. According to the diagram from Fig. 11, such composition range corresponds to liquid phase. Since composition C corresponds to higher liquid fractions than the others, this result might be closer to the liquid in equilibrium with 2223 than the Ca-rich 2201 composition. In fact, the melt in equilibrium with the 2212 phase possibly falls closer to the Ca-rich 2201 primary phase-field, whereas the melt in equilibrium with the 2223 phase might have a 2212-like composition. This becomes clearer by recalling the main solid-state reactions forming 2212 and 2223 by sintering5:

Considering the above reactions in the decomposition direction allows the conclusion that, above the peritectic melting temperatures, 2212 may decompose into a 2201-like liquid, 2:1 and CuO, whereas 2223 would decompose into a 2212-like liquid, 2:1 and CuO. Moreover, the liquid in equilibrium with 2223 could move from the 2212-like composition towards the 2201-like one, becoming richer in Bi by loosing Ca and Cu due to the precipitation of CaCuO phases, as a function of the sample composition, the temperature and the cooling rate. Consequently, a deviation from the 2212-like melt to the 2201-like melt would occur favoring thus the crystallization of 2201 and/or 2212, instead of 2223. Indeed, a slight decrease in the Ca and Cu of the melt with increasing temperature has been shown31.
As long as 2223 exhibits a very limited stability range, the liquid coexisting with this phase might also occupy a very small equilibrium volume surrounded by very flat but elongated multiphase regions pertaining to the Bi-Sr-Ca-Cu-O system. This explains why it is so challenging to find the concentration region where 2223 is the only Bi-Sr-Ca-Cu-O phase in equilibrium with the liquid.
In the present work, 2212 and possibly also 2223 formation have only been indicated for nominal compositions relatively close to the liquid field, whereas the composition with a high excess of Ca and Cu gave lower liquid amounts and too much Ca2CuO3 and CuO. Moreover, neither 2212 nor 2223 formed from melting at an excessively high temperature, which produced many large Ca2CuO3. These results, together with the discussion above, may lead to the conclusion that the melt composition moves from 2212 towards Ca-rich 2201 as the temperature raises, being enriched with Bi and loosing Ca and Cu due to the precipitation of Ca-Cu-O phases. On the other hand, the quenched samples exhibited high Ca-rich 2201 contents, regardless of temperature and composition. We attribute this fact to the high crystallization rate of the 2201 phase. High Bi contents and/or low Sr/Ca ratios tend to promote more liquid formation, favoring thus the precipitation of Ca-rich 2201. On the other hand, the presence of high liquid concentrations favored the formation of long plates with compositions falling in the vicinity of the 2223 primary phase field and might also have formed 2223 or just partially converted the recrystallized 2212 into that phase. The latter assumption seems more feasible, since a reaction path forming firstly 2212 should require lower activation energy. Hence, the formation of large 2212 amounts can be advantageous, rather than an obstacle in promoting the 2223 formation through a subsequent long annealing step. Therefore, our results suggest that long 2223 grains may be achieved, if the optimal precursor composition and treatment conditions can be found.
It has been shown, however, that the formation of relevant 2223 fractions is a difficult task, due to the presence of stable secondary-phases20. High Ca and Cu contents promoted the precipitation and growth of 2:1 in various melting-crystallized samples of the present study. This phase, as well as CuO, have been found as equilibrium compounds in the melting-crystallization treatments, but not acted as appropriate Ca and Cu sources, since they remained and sometimes even grew upon slow-cooling, instead of being consumed to promote 2223 formation. On the other hand, certain amounts of such secondary-phases are important to convert 2212 into 2223 and reach high-critical current densities in sintered tapes41,42. Although it is not a melt-processing, a nucleation and growth model states that the 2212 phase decomposes gradually and locally, reacting with a liquid by which Ca and Cu diffuse to the reaction front forming 2223. In this manner, this reaction can be viewed as a local melt-crystallization, indicating that 2223 may form from a peritectic reaction even in the conventional sintering process. The question arises whether such reaction can occur in the presence of large amounts of liquid, by cooling from temperatures situated above the 2212 and 2223 liquidus lines. Although the reversibility of the 2223 pertitectic melting has been recently shown25, the 2223 phase was not fully decomposed. Indeed, the combination of sintering with a partial 2223 melting step has been succeeded in promoting high texture degrees and critical current densities, being a suitable method for large-scale production of 2223/Ag tapes43-46.
5. Conclusions
The melting and solidification behavior of silver-packed Bi2Sr2Ca2Cu3 O10 (Bi-2223) precursors has been studied. Nominal compositions corresponding to multiphase regions including 2212, Ca2CuO3 (2:1), CuO and liquid have been investigated. A melting-crystallization method, which consists of a partial melting followed by a slow-cooling step, has been applied.
Precursor powders having very distinct nominal compositions presented mainly the Bi2Sr2CaCu2O8 (Bi-2212) phase together with 2:1 and CuO, after sintering at 820840 ºC in air. The silver-packed precursors quenched directly from the melting at 850-900 ºC in 0.08 atm O2 presented 2:1 and CuO grains embedded in a Bi2(Sr,Ca)1.5CuOx (Ca-rich 2201) matrix. Such phase assemblage has been concluded to arise from the peritectic melting of the 2212 phase into liquid and 2:1 taking place within the 2223 compositional range. The Ca-rich 2201 phase crystallizes directly from the melt, possibly even during quenching.
The melt composition probably lay between the Ca-rich 2201 and 2212 stoichiometries. The melt in equilibrium with the 2212 phase may fall closer to the Ca-rich 2201 primary phase-field, whereas the melt in equilibrium with the 2223 phase might have a 2212-like composition. Heating at excessively high temperatures and/or for long times can induce more Ca and Cu segregation from the melt, forming large 2:1 and CuO precipitates. Moreover, too high temperatures and long holding times may induce bismuth and oxygen volatilization, especially for low O2 partial pressures. Consequently, a deviation from the 2212-like melt towards the 2201-like melt may occur, favoring thus the crystallization of 2201 and/or 2212, instead of 2223. Even 2212 could not be recovered by slow-cooling from 900 ºC, but long 2212 plates were able to grow through slow-cooling from 855 ºC, showing that the maximum heating temperature should not be too higher than the melting temperatures. It is possible that some 2223 have also formed, but the limited stability range and sluggish formation kinetic of this phase makes it a challenge to avoid 2201 and 2212 precipitation.
However, regardless the overall composition, the recrystallized 2212 exhibited compositions falling in the vicinity of the 2223 primary phase field. These long 2212 plates can represent a good indication, rather than an obstacle, for the 2223 formation. If a suitable post-annealing is applied, such 2212 is expected to convert more efficiently into long 2223 plates. The main problem regarding such post-annealing method is the presence of high Ca-rich 2201 amounts and large 2:1 and CuO islands tending to remain in their equilibrium condition, rather than react to provide enough Ca and Cu to form 2223. The optimal precursor composition and treatment conditions must be found to form considerable 2223 amounts, either if it occurs directly from a peritectic crystallization or from the conversion of the previously precipitated 2212. With this aim, it is essential to further study and elucidate the 2223-melt equilibrium.
Acknowledgements
A. Polasek would like to thank CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and DAAD (German Academic Exchange Service) for their financial support. We wish also to acknowledge the Pulvermetallurgisches Laboratorium of the Max-Planck-Institut für Metallforschung, in Stuttgart, Germany, where most of the experimental work was done.
Received: June 26, 2003; Revised: May 24, 2004
- 1. Chu, C.W. IEEE Transactions on Appl. Supercond., v. 7, n. 2, p. 80, 1997.
- 2. Larbalestier, D.C. IEEE Transactions on Appl. Supercond., v. 7, n. 2, p. 90, 1997.
- 3. Kitaguchi, H.; Kumakura, H. MRS BULLETIN, v. 26, n. 2, p. 121, February / 2001.
- 4. Flükiger, R.; Grasso, G.; Grivel, J.C.; Marti, F.; Dhallé, M.; Huang, Y. Supercond. Sci. Technol., v. 10, p. A68, 1997.
- 5. Majewski, P. Adv. Mat., v. 6, p. 460, 1994.
- 6. Majewski, P. J. Mater. Res., v. 15, n. 4, p. 854, 2000.
- 7. Roth, R.S.; Rawn, C.J.; Burton, B.P.; Beech, F. J. Res. NIST, v. 95, p.291, 1990.
- 8. Grant, P.M. IEEE Transactions on Appl. Supercond., v. 7, n. 2, p. 112, 1997.
- 9. Feng, Y. et al. Physica C, v. 192, p. 293, 1992.
- 10. Grasso, G.; Hensel, B.; Jeremie, A.; Flükiger, R. Physica C, v. 241, p. 45, 1994.
- 11. Jiang, J. et al., IEEE Trans. on Appl. Supercond., v. 11, n. 1, p. 3561, 2001.
- 12. Cai, X.Y.; Polyanskii, A.; Li, Q., Riley Jr., G.N.; Larbalestier, D.C. Nature, v. 392, p. 30, 1998.
- 13. Wang, W.G.; Horvat, J.; Li, J.N.; Liu, H.K.; Dou, S.X. Physica C, v. 297, p. 1, 1998.
- 14. Lisboa, M.B.; Soares, G.A.; Serra, E.T.; Polasek, A.; Xia, S.K. Materials Characterization, v. 46, p. 75, 2001.
- 15. Buhl, D.; Lang, T.; Cantoni, M.; Risold, D.; Hallstedt, B.; Gauckler, L.J. Physica C, v. 257, p. 151, 1996.
- 16. Marinkovic, B.A.; Xia, S.K.; Saléh, L.A.; Sens, M.; Serra, E.T.; Avillez, R.R.; Rizzo, F.C. Materials Research, v. 5, n. 1, p. 179, 2002.
- 17. Oka, Y.; Yamamoto, N.; Kitaguchi, H.; Oda, K.; Takada, K. Jpn. J. Appl. Phys., v. 28, n. 2, L213, 1989.
- 18. Bock, J.; Preisler, E. Proceedings of ICMC ' 90 Topical-Conference on Materials Aspects of High-Temperature Superconductors, Garmisch-Partenkirchen, Germany, p. 215, 1990.
- 19. Yamada, Y.; Graf, T.; Seibt, E.; Flükiger, R. IEEE Trans. Magn., v. 27, p. 1495, 1991.
- 20. Flükiger, R.; Giannini, E.; Lomello-Tafin, M.; Dhallé, M.; Walker, E. IEEE Trans. Appl. Supercond., v. 11, n. 1, p. 3393, 2001.
- 21. Majewski, P. personal communication.
- 22. Marchetta, M.; Dimesso, L.; Migliori, A.; Masini, R.; Calestani, G. Il Nuovo Cimento, v. 19 D, n. 8-9, p. 1123, 1997.
- 23. Polasek, A.; Rizzo, F.; Serra, E.T.; Majewski, P.; Aldinger, F. presented on MRS Fall Meeting, Materials Research Society, Boston, USA, December / 2002.
- 24. Polasek, A., D.Sc. Thesis, Dept. of Materials Science and Metallurgy, Pontifical Catholic University of Rio de Janeiro - RJ, Brazil, May / 2002.
- 25. Giannini, E.; Passerini, R.; Toulemonde, P.; Walker, E.; Lomello-Tafin, M.; Sheptyakov, D.; Flükiger, R. Physica C, v. 372-376, p. 895, 2002.
- 26. Majewski, P.; Su, H-L; Quilitz, M. J. Mater. Sci. 32, p. 5137, 1997.
- 27. Majewski, P. Supercond. Sci. Technol., v. 10, p. 453, 1997.
- 28. Neves, M.A.; da Silveira, M.F.; Soares, V. Physica C, v. 354, p. 391, 2001.
- 29. Assal, J.; Hallstedt, B.; Gauckler, L.J. Z. Metallkunde, v. 90, n. 12, p. 1025, 1999.
- 30. Majewski, P. Habilitationsschrift zur Erlangung der Venia Legendi für das Fach Mineralogie, Fakultät Bio-und Geowissenschaften, Universität Stuttgart, Deutschland, 1997.
- 31. Styve, V.J.; Geny, J.; Meen, J.K.; Elthon, D. Solid-State Chemistry of Inorganic Materials II, Materials Research Society Proceedings, Boston MA, USA, ed. S. M. McCarron III,A.W. Sleight, and H-C. zur Loye, v. 547, p. 273, 1999.
- 32. Park, C.; Wong-Ng, W.; Cook, L.P.; Snyder, R.L.; Sastry, P.V.P.S.S.; West, A.R. Physica C, v. 304, p. 265, 1998.
- 33. Lu, X.Y.; Nagata, A.; Sugawara, K.; Kamada, S. Physica C, v. 354, p. 313, 2001.
- 34. Schulze, K.; Majewski, P.; Hettich, B.; Petzow, G.; Z. Metallkunde, v. 81, p. 836, 1990.
- 35. Majewski, P.; Hettich, B.; Schulze, K.; Petzow, G. Adv. Mater., v. 3, n. 10, p. 488, 1991.
- 36. Strobel, P.; Tolédano, J.C.; Morin, D.; Schneck, J.; Vacquier, G.; Monnereau, O.; Primot, J.; Fournier, T. Physica C, v. 201, p. 27, 1992.
- 37. Wong-Ng, W. et al., J. Mater. Res., v. 12, n. 11, p. 2855, 1997.
- 38. Lomello-Tafin, M.; Giannini, E.; Walker, E.; Cerutti, P.; Seeber, B.; Flükiger, R. IEEE Trans. Appl. Supercond., v. 11, n. 1, p. 3438, 2001.
- 39. Hatano, T.; Aota, K.; Ikeda, S.; Nakamura, K.; Ogawa, K. Jpn. J. Appl. Phys., v. 27, n. 11, p. L2055, 1988.
- 40. Liu, H. et al., J. Mater. Sci., v. 33, p. 3661, 1998.
- 41. Polasek, A.; Xia, S.K.; Lisboa, M.B.; Sens, M.A.; Serra, E.T.; Rizzo, F.; Borges, H. IEEE Trans. on Appl. Supercond., v.9, n.2, p. 2573, 1999.
- 42. Xia, S.K.; Lisboa, M.B.; Polasek, A.; Sens, M.A.; Serra, E.T.; Rizzo, F.; Borges, H. Physica C, v. 354, p. 467, 2001.
- 43. Dou, S.X.; Liu, H.K.; Guo, Y.C. Appl. Phys. Lett., v. 60, n. 23, p. 2929, 1992.
- 44. Xia, S.K.; Serra, E.T.; Rizzo, F. Physica C, v. 361, p. 175, 2001. Continuous Cooling Process (CCS), patent pending.
- 45. Xia, S.K.; Lisboa, M.B.; Serra, E.T.; Rizzo, F. Supercond. Sci Technol., v. 14, p. 103, 2001.
- 46. Xia, S.K.; Serra, E.T. Studies of High Temperature Superconductors, ed. A. Narlikar, Nova Science Publisher, New York, USA, v. 43, p. 63, 2002.
Publication Dates
-
Publication in this collection
13 Aug 2004 -
Date of issue
Sept 2004
History
-
Received
26 June 2003 -
Reviewed
24 May 2004