Resumo
Thalassemias are a heterogeneous group of inherited disorders characterized by a microcytic hypochromic anemia and an imbalance in the synthesis of the globin-chains. Hb C is the second most frequently variant of hemoglobin found in Brazil. The laboratory diagnosis of hemoglobinopathies, including thalassemias, is growing in importance, particularly because of an increasing requirement for neonatal diagnosis of abnormal hemoglobins. Screening tests were carried out using alkaline and acid electrophoresis, globin-chain analysis by cellulose acetate in alkaline pH, isoelectric focusing and HPLC. The molecular characterization was made by PCR-ASO for Hb C and beta thalassemia mutants. Large-scale screening and discriminative methodologies must provide information about the hemoglobin polymorphisms in Brazilian population. HPLC is a powerful tool in these cases. Molecular characterization is important to genetic counseling and clinical management, in particular for the Brazilian population that have an intense racial admixture, with great variability of hemoglobins. In this paper an association between Hb C and beta thalassemia (IVS-II-654) in a black family from Brazil was described.
CARTA AO EDITOR
Interação entre Hb C [beta6(A3)Glu>Lys] e IVS II-654 (C>T) beta-talassemia no Brasil
Hb C [beta6(A3)Glu>Lys] and IVS II - 654 (C>T) beta thalassemia interaction in Brazil
Claudia R. Bonini-DomingosI; Ana C. Bonini-DomingosII; Ana R. ChinelatoIII; Paula J. A. ZamaroIV; Patrícia H. O. CalderanV
ILHGDH-IBILCE-UNESP, São José do Rio Preto, SP
IIGraduanda do Curso de Enfermagem da UNIP, São José do Rio Preto, SP; estagiária de iniciação científica do LHGDH-IBILCE-UNESP, São José do Rio Preto, SP
IIIDoutoranda do LHGDH-IBILCE-UNESP, São José do Rio Preto, SP
IVMestranda do LHDGH-IBILCE-UNESP, São José do Rio Preto, SP
VProfessora responsável pelo Laboratório de Hematologia UNIC, Cuiabá, MT
Endereço para correspondência Endereço para correspondência Claudia R. Bonini-Domingos LHGDH-IBILCE-UNESP Rua Cristóvão Colombo, 2.265 Jd. Nazareth 15054-000 São José do Rio Preto-SP Brasil Tel: (17) 221-2392 Fax: (17) 221-2390 E-mail: claudiabonini@yahoo.com.br
ABSTRACT
Thalassemias are a heterogeneous group of inherited disorders characterized by a microcytic hypochromic anemia and an imbalance in the synthesis of the globin-chains. Hb C is the second most frequently variant of hemoglobin found in Brazil. The laboratory diagnosis of hemoglobinopathies, including thalassemias, is growing in importance, particularly because of an increasing requirement for neonatal diagnosis of abnormal hemoglobins. Screening tests were carried out using alkaline and acid electrophoresis, globin-chain analysis by cellulose acetate in alkaline pH, isoelectric focusing and HPLC. The molecular characterization was made by PCR-ASO for Hb C and beta thalassemia mutants. Large-scale screening and discriminative methodologies must provide information about the hemoglobin polymorphisms in Brazilian population. HPLC is a powerful tool in these cases. Molecular characterization is important to genetic counseling and clinical management, in particular for the Brazilian population that have an intense racial admixture, with great variability of hemoglobins. In this paper an association between Hb C and beta thalassemia (IVS-II-654) in a black family from Brazil was described.
Senhor Editor
A hemoglobina C (Hb C) é uma variante originada pela substituição do ácido glutâmico por lisina na posição 6 da betaglobina, causando um leve distúrbio hemolítico. Possui prevalência entre 15% a 30% nos povos de origem africana, e sua freqüência é bastante variável na população brasileira, dependendo da região analisada. A troca do aminoácido confere características estruturais e funcionais próprias à molécula, facilitando a sua identificação por metodologias de rotina diagnóstica.1 A co-migração eletroforética com outras variantes de hemoglobina pode ser elucidada pela utilização de metodologias mais sensíveis, como a cromatografia líquida de alta pressão (HPLC). Uma característica particular da população brasileira reside na miscigenação, facilmente observada pela herança de diferentes formas de hemoglobinas anormais em associação. Caracterizar os mutantes de hemoglobinas em núcleos familiares com utilização de metodologias de rotina e biologia molecular passou a ser importante para o direcionamento clínico e orientação aos portadores.2
Estudaram-se amostras de sangue de indivíduos de um núcleo familiar do interior do estado do Mato Grosso, de ascendência negróide, segundo os relatos familiares. Foram colhidas amostras de sangue periférico após consentimento, acondicionadas em tubos com EDTA como anticoagulante. Os experimentos foram realizados no Laboratório de Hemoglobinas e Genética das Doenças Hematológicas LHGDH, Departamento de Biologia, do Instituto de Biociências, Letras e Ciências Exatas IBILCE-UNESP de São José do Rio Preto, SP, Brasil. As amostras de sangue foram triadas por metodologias de rotina como: teste de resistência globular osmótica em solução de cloreto de sódio a 0,36%,3 metodologia utilizada como teste seletivo para talassemias; análise da morfologia eritrocitária;2 eletroforese em pH alcalino.4 A presença das hemoglobinas anormais foi confirmada por testes complementares, tais como: eletroforese em pH ácido;5 isoeletrofocalização;6 eletroforese de cadeias polipeptídicas;7 HPLC utilizando-se o equipamento VARIANT da BIO-RAD e kit diagnóstico betatalassemia heterozigota, seguindo protocolo do fabricante.
Para a confirmação dos mutantes de hemoglobinas, foi feita a análise molecular por amplificação e hibridação com sonda Oligonucleotídeo Alelo Específico (ASO) para hemoglobinas S e C e para os mutantes que determinam betatalassemia, utilizando-se os kits de diagnóstico da linha mDx-BIO-RAD, seguindo protocolo do fabricante.
A hemoglobina C é freqüente entre povos da África, onde a prevalência da heterozigose (Hb AC) para essa hemoglobina atinge 30% da população. Os portadores heterozigotos são assintomáticos, enquanto o estado de homozigose (HbCC) é caracterizado por anemia hemolítica de intensidade variável. A origem da hemoglobina C, tal como da hemoglobina S, é africana e sua propagação foi ampla na região do Mediterrâneo e Américas por meio do tráfico de escravos, em diferentes períodos da história da humanidade. Esse processo de distribuição do gene da globina beta possibilitou sua interação com outras hemoglobinas variantes e talassemias, amplamente observado na população brasileira.
O propósito do núcleo familiar em estudo foi uma mulher de 47 anos, de origem negróide, com anemia discreta. A procura por serviços especializados fundamentou-se na presença de anemia de longa data, sem resultado aos tratamentos convencionais e com diagnóstico anterior de Hb C em homozigose.
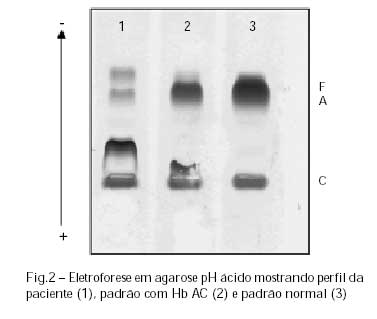
Nos procedimentos de triagem realizados observou-se resistência osmótica em solução de NaCl a 0,36% positiva, morfologia eritrocitária com moderada microcitose, hipocromia e presença de células em alvo. Nos procedimentos eletroforéticos em pH alcalino observou-se padrão de migração semelhante à hemoglobina C em homozigose (Figura 1). Frações na posição de hemoglobina A e Hb F em pequena concentração, além de hemoglobina C desviada da posição normal, foram observadas na eletroforese em pH ácido (Figura 2), confirmados por focalização isoelétrica, como teste complementar à triagem inicial. A eletroforese de globinas mostrou presença de cadeias alfa A e grande quantidade de mutante na posição da cadeia beta C (Figura 3). O conjunto das análises eletroforéticas sugeria Hb CC.
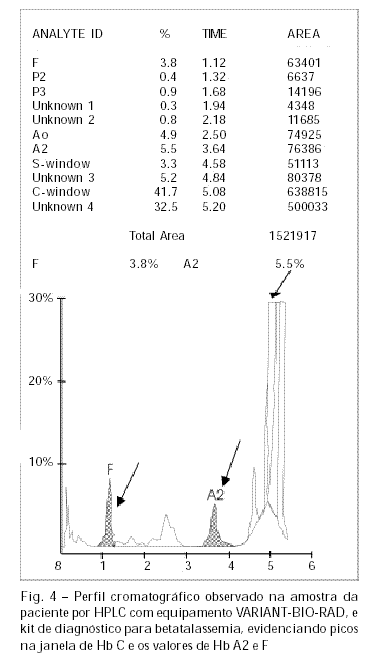
As análises por HPLC mostraram cromatograma com alguns picos desconhecidos e duplo pico na janela de hemoglobina C, sendo a concentração de 41,7% para Hb C e de 32,6% para uma hemoglobina desconhecida sugerindo interação de hemoglobina C com outro mutante de cadeia beta (Figura 4). A fração correspondente à Hb A apresentou-se com concentração muito baixa, de 4,9 %. As Hb F e Hb A2 apresentaram valores de 3,8% e 5,5% respectivamente.
Os valores de Hb A2 e F foram confirmados pelos métodos de eluição para Hb A2 e desnaturação alcalina para Hb F, sendo compatíveis com os observados por HPLC.
As análises de triagem e confirmação realizadas nas amostras de sangue das filhas da paciente mostrou, em conjunto, fenótipo de talassemia do tipo beta heterozigota, com valores de Hb A2 e fetal discretamente aumentados e hemograma com valores no limite inferior de normalidade.
O conjunto dos testes eletroforéticos e hematológicos, aliados às análises cromatográficas por HPLC foram decisivos para a pesquisa do mutante por biologia molecular. Na avaliação da presença dos mutantes por PCR-ASO, utilizando-se o kit mDx Hb S/ C da BIO-RAD, o padrão observado para a paciente foi de heterozigoto para hemoglobina C, não compatível com os resultados eletroforéticos, mas sugerindo a interação de hemoglobina C com outro mutante, não identificado, no entanto, por essa metodologia.
Como as filhas expressavam fenótipo de talassemia do tipo beta em heterozigose, rastrearam-se os oito mutantes mais freqüentes de origem mediterrânea pelo kit mDx beta gene 1, os quais se mostraram todos negativos. A presença da mutação IVS II: 654 na paciente foi detectada através do kit mDx beta gene 2, que possui sondas para os oito mutantes mais freqüentes de origem asiática. Duas filhas apresentaram heterozigose para os seguintes mutantes de talassemia beta: IVS II-654 e CD 17; uma outra filha apresentou interação entre IVS II-654 e CD 17.
O pai não realizou exames nesta ocasião e foi relatado como portador de hemoglobinas normais. Com a interação observada em uma das filhas e a presença de mutante diferente em outra, o pai deverá ser reavaliado. Os resultados foram informados à família em sessão de aconselhamento genético e repassados ao clínico que acompanha a paciente, elucidando as dúvidas diagnósticas e facilitando o tratamento.
Os mutantes de hemoglobina observados na família em estudo, descritos pela primeira vez no Brasil, refletem a intensa miscigenação da população brasileira e ressaltam a importância da utilização de testes complementares com boa resolução no diagnóstico das hemoglobinas, em especial nos casos de interações, fornecendo informações importantes para a conduta terapêutica e orientação genética.
A mutação IVS-II-654 (C>T); AAGGCAATA>AAG^GTAATA para betatalassemia é encontrada em chineses e descendentes de asiáticos. A mutação CD17 (A>T) AAG(Lys)->TAG para betatalassemia cria um códon de terminalização precoce, afetando a tradução.8 Neste caso encontrou-se a mutação em uma família de origem negróide sem relatos de ascendência asiática.
Recebido: 28/07/02
Aceito: 14/01/03
Trabalho desenvolvido no Laboratório de Hemoglobinas e Genética das Doenças Hematológicas, IBILCE, UNESP, São José do Rio Preto, SP.
- 1. Old J. Haemoglobinopathies. Prenatal Diagnosis 1966; 16:1181-1186.
- 2. Bonini-Domingos CR. Hemoglobinopatias no Brasil: variabilidade genética e metodologia laboratorial. São José do Rio Preto, 1993. Tese (Doutoramento em Ciências Biológicas) Instituto de Biociências Letras e Ciências Exatas, Universidade Estadual Paulista.
- 3. Silvestroni E, Bianco I. Screening for microcytemia in Italy: analysis of data collected in the past 30 years. Am J Hum Genet 1975;27:198-212.
- 4. Marengo-Rowe AJ. Radip electrophoresis and quantitation fo haemoglobin on cellulose acetate. J Clin Path 1965;18:790-792.
- 5. Vella F. Acid agar gel electrophoresis of human hemoglobin. Am J Clin Path 1968;49:440-442.
- 6. Naoum PC. Eletroforese, técnicas e diagnósticos. São Paulo: Santos, 1999.
- 7. Schneider RG. Differentiation of electrophoretically hemoglobins suchas S, D, G and P or A2, C, E, and O by electrophoresis of the globin chains. Clin Chem 1974;20:1111-5.
- 8. http://globin.cse.psu.edu/
Endereço para correspondência
Datas de Publicação
-
Publicação nesta coleção
09 Dez 2003 -
Data do Fascículo
Jun 2003
Histórico
-
Aceito
14 Jan 2003 -
Recebido
28 Jul 2002