Abstract
Salmonella enterica and Shigella species are commonly associated with food and water borne infections leading to gastrointestinal diseases. The present work was undertaken to develop a sensitive and reliable PCR based detection system for simultaneous detection of Salmonella enterica and Shigella at species level. For this the conserved regions of specific genes namely ipaH1, ipaH, wbgZ, wzy and invA were targeted for detection of Shigella genus, S. flexneri, S. sonnei, S. boydii and Salmonella enterica respectively along with an internal amplification control (IAC). The results showed that twenty Salmonella and eleven Shigella spp., were accurately identified by the assay without showing non-specificity against closely related other Enterobacteriaceae organisms and also against other pathogens. Further evaluation of multiplex PCR was undertaken on 50 natural samples of chicken, eggs and poultry litter and results compared with conventional culture isolation and identification procedure. The multiplex PCR identified the presence of Salmonella and Shigella strains with a short pre-enrichment step of 5 h in peptone water and the same samples were processed by conventional procedures for comparison. Therefore, this reported multiplex PCR can serve as an alternative to the tedious time-consuming procedure of culture and identification in food safety laboratories.
Salmonella; Shigella; multiplex PCR; spiking
A novel multiplex PCR for the simultaneous detection of Salmonella enterica and Shigella species
M. Radhika; Majumder Saugata; H.S. Murali; H.V. Batra
Microbiology Division, Defence Food Research Laboratory, Siddharthanagar, Mysore, Karnataka, India
Send correspondence to Send correspondence to M. Radhika Microbiology Division, Defence Food Research Laboratory Siddharthanagar, 570011 Mysore, Karnataka, India E-mail: mursradhika@yahoo.co.in
ABSTRACT
Salmonella enterica and Shigella species are commonly associated with food and water borne infections leading to gastrointestinal diseases. The present work was undertaken to develop a sensitive and reliable PCR based detection system for simultaneous detection of Salmonella enterica and Shigella at species level. For this the conserved regions of specific genes namely ipaH1, ipaH, wbgZ, wzy and invA were targeted for detection of Shigella genus, S. flexneri, S. sonnei, S. boydii and Salmonella enterica respectively along with an internal amplification control (IAC). The results showed that twenty Salmonella and eleven Shigella spp., were accurately identified by the assay without showing non-specificity against closely related other Enterobacteriaceae organisms and also against other pathogens. Further evaluation of multiplex PCR was undertaken on 50 natural samples of chicken, eggs and poultry litter and results compared with conventional culture isolation and identification procedure. The multiplex PCR identified the presence of Salmonella and Shigella strains with a short pre-enrichment step of 5 h in peptone water and the same samples were processed by conventional procedures for comparison. Therefore, this reported multiplex PCR can serve as an alternative to the tedious time-consuming procedure of culture and identification in food safety laboratories.
Key words:Salmonella, Shigella, multiplex PCR, spiking.
Introduction
Diarrhoeal disease is an important public health problem in developing countries and also is an important cause of morbidity and mortality throughout the world. Globally Salmonella enterica and Shigella species remain major contributors to acute enteric infections (Jousilahti et al., 1997). They are closely related enteric pathogens belonging to the family Enterobacteriaceae and are found naturally in the environment, humans and food (Jun et al., 2004). Bacillary dysentery or Shigellosis caused by Shigella spp., is endemic throughout the world and it is among the most common causes of bacterial diarrhoeal diseases. It is responsible for approximately 165 million cases annually, of which 163 million are in developing countries and 1.5 million in industrialized ones (Kumar et al., 2010). Shigella spp., are subdivided into four serogroups - S. sonnei, S. boydii, S. flexneri and S. dysenteriae and humans are the principal reservoir of infection. The infectious dose of Shigella is as low as 10 bacterial cells (Germani and Sansonetti, 2006) and the transmission occurs person to person through the faecal-oral pathway, and this feature of shigellosis gives the pathogen epidemic outbreak potential and can rapidly be disseminated through contaminated food and water (Niyogi, 2005). The symptoms of Shigella infection range from mild watery diarrhoea normally in case of S. sonnei to severe bacillary dysentery with fever, abdominal pain, blood and mucus in stool samples caused mainly by strains of S. dysenteriae 1 (Kumar et al., 2006). S. flexneri and S. boydii can cause either mild or severe illnesses.
Salmonella enterica are a major cause of foodborne illness leading to acute gastroenteritis throughout the world. Salmonella infections (Salmonellosis) are transmitted from animals to humans through food and occasionally from person to person through the fecal-oral route (D'Aoust, 1989). Although gastroenteritis often occurs in large epidemics among individuals who have eaten contaminated food, family outbreaks and sporadic cases are even more common (Moffat et al., 2012). Salmonellosis in the human occurs in a variety of forms, presenting a broad clinical spectrum. Salmonella infections of humans and animals continue to remain a major public health hazard worldwide. This also has a large negative economic impact on food production.
With the continuing impact of these pathogens on human and animal health, the need for rapid and accurate detection methods for these organisms both in environmental and clinical samples remains high. Moreover, identification of Salmonella enterica and Shigella species is important for surveillance, prevention and control of food-borne diseases (Li et al., 2005). An accurate and rapid procedure for identification would help identify the sources, reservoirs and or their transfer through food chain. Conventional methods recommended for the detection of these pathogens are generally cumbersome, inefficient, labor intensive and time consuming (Miescier et al., 1992; D'Aoust, 1989). DNA-based methods are better for identification of food-borne pathogens because these methods rely on the nucleic acid composition of the bacterium instead of their phenotypic expressions that may be variable under culture conditions (Hill et al., 1985; Olsen et al., 1995; Batt, 1997). The PCR represents a rapid procedure with both high sensitivity and specificity for the immediate detection, identification of specific pathogenic bacteria from various food matrices (Lantz et al., 1994; Hill, 1996) and multiplex PCR; has been used to detect several pathogens in a single reaction (Way et al., 1993; Brasher et al., 1998; Soumet et al., 1999).
Multiplex PCRs have been reported where both Salmonella enterica and Shigella genus could be identified together but not with the individual species (Vantarakis et al., 2000; Kong et al., 2002; Li and Mustafa, 2004). In few multiplex PCR reports the targeted genes such as Vir A and invA (Vantarakis et al., 2000; Villalobo and Torres, 1998), ipaH (Kong et al., 2002), phoP/phoQ (Li and Mustafa, 2004), were not totally specific as some E.coli genes were also getting amplified. Therefore, it was planned to work out on a multiplex PCR that can conclusively differentiate all four Shigella spp., along with Salmonella enterica and that could be directly applied for the food samples. The new multiplex PCR system standardized in this study could detect target organisms directly from food and environmental samples with 5 h preenrichment step and without exhibiting non specific amplification even among closely related organisms. The genomic DNA could be extracted by boiling method in order to save time and also to reduce the cost. The sensitivity of the newly developed multiplex PCR is better when compared with the reported (Vantarakis et al., 2000) multiplex PCR.
For this multiplex PCR two genus specific genes invA (Invasion protein gene 'A') of Salmonella enterica (Rahn et al., 1992) and ipaH1 (invasion plasmid antigen H) gene of Shigella (Brigittle et al., 1998) were selected. For species differentiation of Shigella, species specific genes wzy (O- antigen polymerase), wbgZ (putative epimerase/dehydratase) and ipaH1 (invasion plasmid antigen H) of S. boydii (Tao et al., 2004), S. sonnei (James et al., 2000) and S. flexneri (Kenia et al., 2010), respectively were selected.
Moreover, an IAC, which has now become almost mandatory for diagnostic PCR's, was also incorporated to account for the false negative results during the PCR reaction. Following standardization, the multiplex PCR was evaluated with Salmonella enterica and Shigella strains individually and in combination to assess the sensitivity and the specificity. Spiking studies were also conducted in food samples to evaluate the efficacy of the system. The detection of all these pathogenic bacteria simultaneously from food samples could be completed within 8-10 h.
Materials and Methods
Bacterial strains and cultural methods
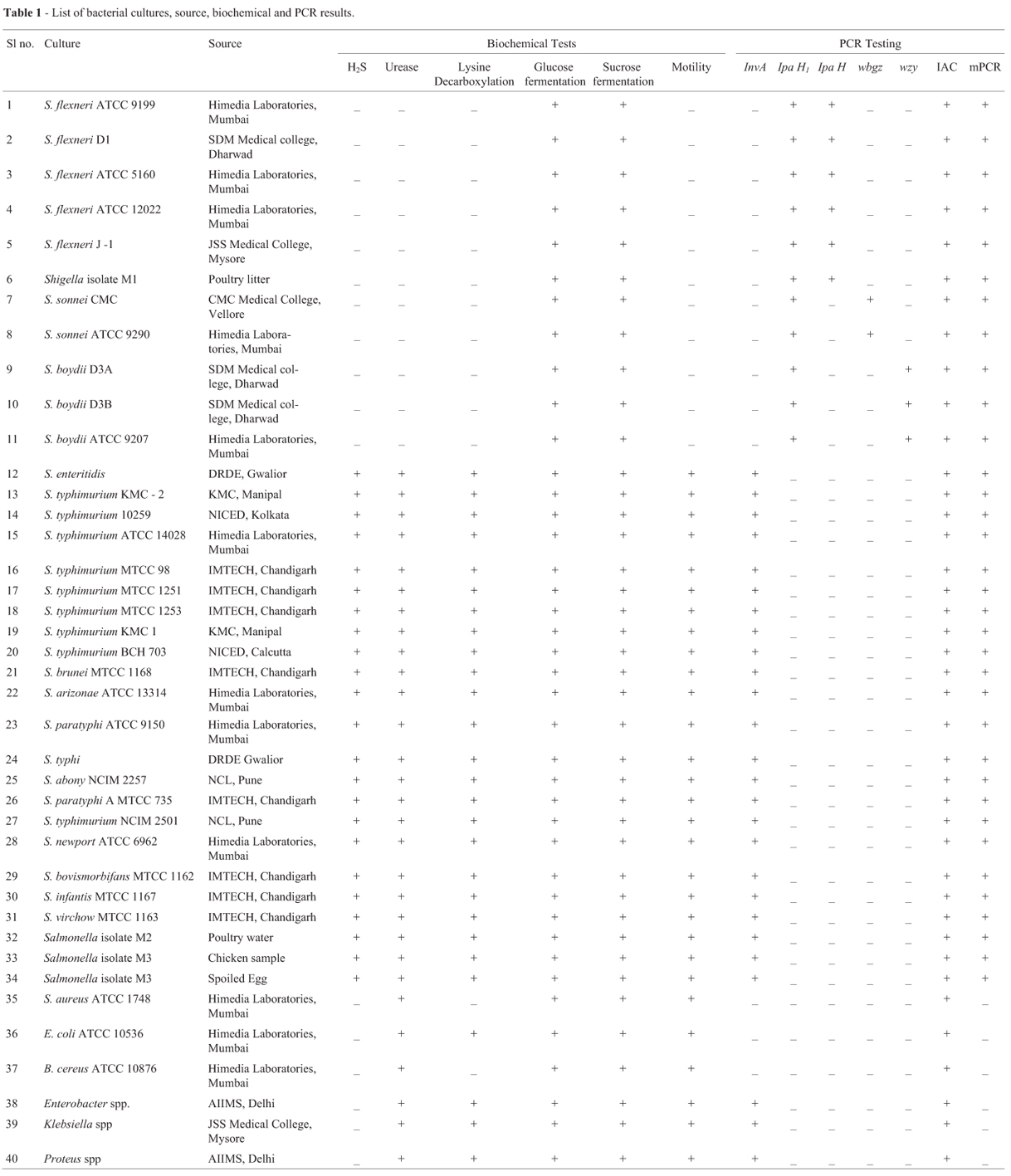
The bacterial strains selected to test the diagnostic specificity of PCR are listed in Table 1. These included reference strains, human clinical isolates and food and environmental isolates. Standard strains and isolates were grown in Nutrient broth and then streaked on selective agars procured from Himedia (Mumbai, India). Presumptive colonies selected on Deoxycholate Citrate Agar (DCA), Salmonella Shigella Agar (SSA) and Xylose Lysine Deoxycholate Agar (XLD) were further subjected to biochemical tests viz., Lysine Decarboxylation, Hydrogen di sulphide (H2S) production, carbohydrate fermentation and Urease production and motility test (Food and Drug Administration, 1995). The bacterial strains were periodically subcultured and were used for DNA extraction.
DNA Extraction
Each tube containing 5 mL of Luria Bertani broth (LB) was inoculated with each culture and incubated (37 ºC, 24 h) in shaking conditions. Template DNA from each bacterial strain was extracted by boiling lysis method (Theron et al., 2001) or by commercial DNA extraction kit (Qiagen, Germany). One milliliter of cells was removed and centrifuged at 10,000 rpm for 3 min. The pellet was resupended in 50 mL of distilled water, boiled for 12 min, and centrifuged again for 3 min at 12,000 rpm. The supernatant was transferred to a new microfuge tube and the DNA concentration was estimated by NanoDrop-ND-1000 spectrophotometer (Thermo scientific, India) and the DNA was stored at -20 ºC until further use.
Primers and Internal amplification control
Five sets of primers were designed to detect invA, ipaH, ipaH1 and wbgZ and wzy genes using Gene Bank database sequences. Conserved regions were selected and primers were designed with Gene runner software (http://www.generunner.com). The gene bank accession numbers of the gene sequences used to design the primers are given in the Table 2. Sequences of all primers (Table 2) were evaluated using the Primer-BLAST and BLASTn tools (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) to identify any non-specific targets and to anticipate the specificity of the PCR assay. PCR products ranged from 254 bp to 866 bp in length. Appropriate care was taken to ensure that the primer pairs had similar annealing temperature and the resulting amplicons had a minimum 50 bp difference among themselves (Table 2). The primer pairs were custom synthesized from Eurofins Pvt Ltd., Bangalore (Table 2).
To check the presence of inhibitors within PCR mixture, an IAC was constructed. The primers used in this reaction had 5' overhanging ends, which were identical to the primer wbgZ (factor essential for putative epimerase/dehydratase protein) whereas 3' ends were complementary to a DNA sequence of pUC 19 (Table 2). The PCR reaction mixture for generation of IAC DNA contained 6 picomolar of each primer, 100µM each dNTPs (Sigma), 0.5 units of Taq polymerase, 2.0 millimolar MgCl2 in 1x PCR buffer (MBI Fermentas, Canada) with 175 ng pUC 19 plasmid DNA was used as template. The reaction procedure consisted of 30 cycles of denaturation at 94 ºC for 1 min, primer annealing at 57 ºC for 1 min and extension at 72 ºC for 1 min. The DNA was denatured for 4 min in the beginning and finally extended for 10 min at 72 ºC (Eppendorf master cycler gradient, Hamburg Germany). PCR product was purified using commercially available kit (Qiagen, Hilden, Germany). The concentration of IAC DNA was determined spectrophotometrically at 260 nm and was stored in DDW at -20 ºC. The following equation was used to calculate the copy number of the PCR product concentration: weight of PCR fragment (in g µL-1) x (6.023 x 1023) / (660 g mol L-1 x number of base pairs of PCR fragment) = the number of genomic copy per microlitre (Mahesh et al., 2009).
Standardization of multiplex PCR
Initially, presence of all genes in the standard strains was confirmed by performing monoplex PCRs. Multiplex PCR was standardized by empirically varying critical factors that affect multiplexing, primer concentrations, amount of MgCl2, annealing time and temperature. Multiplex PCR was optimized in an Eppendorf master thermal cycler (Hamburg, Germany) with a reaction volume of 30 µL reaction containing 2.5 picomolar each of IpaH1 F and IpaH1 R primers, 4.0 picomolar each of wzy F and wzy R primers, 5.0 picomolar each of IpaH F and IpaH R primers and 6.0 picomolar each of wbgZ and invA F and wbgZ and invA R primers, 200 µM of each dNTP, 7000 copies of IAC DNA, 1 unit of Taq polymerase, 2 mM MgCl2 in 1x PCR buffer with 1.2 L of mixed template DNA from bacterial samples. Amplification consisted of initial denaturation at 94 ºC for 4 min followed by 30 cycles of denaturation at 94 ºC for 1 min, primer annealing at 57 ºC for 1 min and extension at 72 ºC for 1 min and followed by a final extension at 72 ºC for 10 min. The PCR products were electrophoresed on 2% agarose gel stained with ethidium bromide and visualized under UV transilluminator (G-Box, syngene, India).
Determination of specificity and sensitivity of multiplex PCR
The specificity of the multiplex PCR primers was determined against different organisms as shown in Table 1 by taking 1.5 µL of template DNA from each of the organism (approx. 107 cfu). The procedure for PCR was essentially the same as described earlier. To assess sensitivity, minimum amount of DNA detectable by multiplex PCR, 10-fold serial dilutions of overnight growth of S. flexneri ATCC 9199, S. sonnei ATCC 9290, S. boydii ATCC 9207 and S. typhimurium MTCC 98 were serially diluted in sterile saline (0.8%) to achieve concentrations of 108-100 cfu/mL. DNA was prepared by boiling method and multiplex PCR assay was performed on the boiled lysates of each diluted culture in order to detect the minimum number of cells. Escherichia coli genomic DNA was used as a negative control.
Analysis of experimentally spiked food sample
In order to estimate the interference of food matrices on the sensitivity, detectable concentration of target organisms (108 cfu/mL) were ten-fold serially diluted in 10 mL saline. Retort processed chicken pulav rinse was prepared by macerating 10 g of sample in 90 mL of peptone water using stomacher. The rinse was centrifuged at 1,500 g to get rid of the particulate debris and supernatant was stored in aliquots of 10 mL at -20 ºC for further use. Ten milliliter each of chicken pulav rinse were spiked with S. flexneri ATCC 9199, S sonnei ATCC 9290, S. boydii ATCC 9207 and S. typhimurium MTCC 98 culture with cell concentration ranging from 108 to 101 cfu mL-1 and incubated overnight (18 h) at 37 ºC. One milliliter was taken at the end of the incubation period from all the samples and processed for DNA extraction by boiling method. The DNA (1.5 µL) was used as template in PCR assay. Simultaneously 100 µL sample from each dilution was plated to assess the cfu's. Similar sampling and analysis were done after 5 h incubation also.
Investigation of Salmonella enterica and Shigella spp., from food and environmental sources
Fifty food and environmental samples obtained from various localities in Mysore region were assessed for the presence of Salmonella enterica and Shigella species following standard enrichment and isolation procedures and also by multiplex PCR method (Table 3). For this 25 g of sample was aseptically cultured in 225 mL of Lactose broth and incubated at 37 ºC for 24 h. About 1 mL of sample was transferred to 9 mL of tetrathionate broth (Himedia, India) and incubated at 37 ºC for 24 h. Subsequently, a loopful of broth was streaked on surface of SDA, SSA and XLD agar (Himedia, India) and incubated at 37 ºC for 24 h. Further, biochemical tests were carried out by conventional methods for suspected isolates (Wallace and Thomas, 1998). Genomic DNA (1.5 µL) prepared from 1 mL enrichment broth (short enrichment incubation step of 5 h in peptone water) by boiling lysis method was used in multiplex PCR.
Results
Bacterial strains and cultural methods
All the cultures as listed in Table 1 were characterized by conventional method and colonies selected on DCA, SSA and XLD agar were further confirmed by biochemical tests. Salmonella cultures were found to be positive for fermentation of Mannitol, Dextrose, Lysine decarboxylation, H2S production, and negative for fermentation of Lactose, Sucrose and urease whereas Shigella cultures were nonmotile, positive for glucose fermentation, negative for H2S, gas production, lysine decarboxylation and urease as shown in (Table 1).
Standardization of Multiplex PCR for the detection of selected genes:
The reaction conditions for the multiplex PCR assay were optimized to ensure that all of the target gene sequences were satisfactorily amplified. Care was taken to avoid areas of homology with other organisms. The concentration of primers was optimized after standardizing individual monoplex PCRs (Figure 1) with their respective genes and had almost equal annealing temperature, which reduced the non-specific amplification The annealing temperature of 57 ºC was selected because at this temperature adequate resolution of all the amplified products could be seen. Serial dilution of IAC incorporated in the multiplex assay revealed that 3000 copies of the same resulted in unhindered amplification of wbgZ gene for which the IAC primer pair was competitive besides meticulously amplifying other multiplex genes (Figure 2) and the detection limit of the assay was found to be 5 pg. The ability of the multiplex PCR to identify Salmonella enterica and Shigella spp., was validated using Salmonella and Shigella spp., and other strains listed in Table 1.
All the cultures were identified correctly by the standardized multiplex PCR (Figure 1 and Table 1). The standardized multiplex PCR revealed the presence of five bands after agarose gel electrophoresis. On amplification, multiplex PCR yielded a band of 780 bp invA gene for Salmonella enterica, 254 bp ipaH1 gene for Shigella genus, 305 bp wzy gene for S. boydii, 460 bp wbgz gene for S. sonnei and 595 bp ipaH gene for S. flexneri on agarose gel electrophoresis. IAC, was coamplified with wbgz primers and had the amplicon size of 866 bp. As a negative control multiplex PCR was tested with water, no amplicons were observed except IAC. This internal control is highly recommended when PCR-based methods are used to detect bacterial pathogens (Hoorfar et al., 2004). All these results were compared with the conventional culture, isolation and biochemical identification procedures. In the multiplex PCR described here, 1.2 x 103 copies of IAC DNA appeared suitable in avoiding competition between target DNA and IAC DNA for wbgz primers particularly at low concentration of target DNA. In PCR with an IAC, a control signal will be produced even when there is no target sequence present. This helps in revealing failures of PCR reactions (Hoorfar et al., 2004).
Robustness
There was no significant loss in the visibility of bands at less (10%, 20%) or more (10%, 20%) concentrations of PCR reagent and IAC DNA. Temperature variation of ± 2 ºC also did not make any appreciable change in the profile.
Specificity and sensitivity of multiplex PCR
There was no amplified product obtained except that of IAC with non Salmonella and Shigella strains (Table 1). All the genes specific for Shigella spp., were detected until 10-8 dilution which was equivalent to 4 x 102 cfu/mL. In case of Salmonella enterica, inv A gene was detected until 10-7 dilution which was equivalent to 2 x 103 cfu/mL.
Analysis of artificially contaminated chicken samples:
The detection limit was found to be 200 cfu/g, 10 cfu/g, and 30 cfu/g and 20 cfu/g of S. typhimurium, S. flexneri, S. sonnei and S. boydii respectively in chicken pulav rinse after 5 h of enrichment (Table 3).
Analysis of field samples
Further evaluation of multiplex PCR was undertaken on 50 natural samples (chicken, eggs and poultry litter) and the comparison done with conventional culture isolation and identification procedure. The multiplex PCR could identify the presence of Salmonella and Shigella in three and one samples respectively and the same samples also yielded target organisms by the conventional method.
Discussion
Salmonella and Shigella spp. are frequently encountered as food borne outbreaks or occurring as sporadic cases (Alejandro et al., 2012). The conventional methods of isolation and identification of these pathogens primarily depends on selective media followed by a battery of biochemical tests that are laborious, costly and time consuming (Radji et al., 2010). Generally, other molecular-based methods like PCRs in monoplex and multiplex formats (Liu et al., 2008), PCR non radioactive labelling, PCR -RFLP PCR -ELISA (Ojha et al., 2013) and DNA microarray have a well grounded potential to overcome certain insufficiencies of the conventional methods and is found to be sensitive and reproducible but its application in diagnostic field is difficult due to its high cost and the requirement of skilled person (Rahn et al., 1992). Several reported PCR do not discriminate between Shigella at the genus and species levels, nor did they differentiate Shigella from closely related pathogens such as Salmonella, Citrobacter and enteroinvasive E. coli (Germani and Sansonetti, 2006; Liu et al., 2008, Vantarakis et al., 2000).
In some multiplex PCR reports, the targeted genes such as Vir A and invA (Vantarakis et al., 2000; Villalobo and Torres, 1998), ipaH (Kong et al., 2002), phoP/phoQ (Li and Mustafa, 2004), were not totally specific as some E.coli genes were also getting amplified. According to the ohja et al. (2013) the primers invC, rfc, wbgz and rfpB designed for the identification of Shigella spp., S. flexneri, S. sonnei and S. dysenteriae were specific however according to NCBI blast analysis the primers showed cross reactivity with other members of enterobacteriaceae family. Most of the reported multiplex PCRs lack the inclusion of an IAC, to rule out false negative reactions (Liu et al., 2008; Vantarakis et al., 2000). The false negative PCR reaction might occur due to variation in performance of PCR thermal cyclers, incorrect PCR mixture, inefficiency in Taq polymerase, personnel and the presence of PCR inhibitors in the sample matrices, therefore inclusion of an IAC in the multiplex PCR becomes necessary (Malorny et al., 2003).
In the current study, we developed a novel multiplex PCR by combining genus specific and species specific genes and also the conserved genes of respective organisms which has enabled the system to identify Salmonella enterica and differentiate Shigella species. In addition, to make the multiplex PCR assay acceptable to the present norms of a diagnostic PCR, IAC was also included. The proficiency of individual primers to yield specific amplification for the target bacterial species was confirmed initially by monoplex PCR's and then adapted into multiplex format. Primers designed for this multiplex PCR were chosen to maintain a near uniform annealing temperature and also care was taken to maintain 50 bp differences among different PCR products for clear resolution. The selected primers were tested to examine the possible cross-reactions of primers by homology searches using the basic local alignment searching tool (BLAST) program. The IAC was incorporated in pUC 19 plasmid DNA flanked by wbgz primers. Optimum number of copies of IAC DNA was selected to give a good visible band in order to avoid competition between target DNA and IAC DNA for wbgz primers. Another important criterion for a diagnostic PCR is robustness. No significant loss in the visibility of bands could be observed even with the variation of PCR reagents as well as IAC and DNA concentration.
The invA gene of Salmonella enterica selected in the study has already been proven to be an important diagnostic tool for detection as it contains sequence unique to this genus (Rahn et al., 1992; Shanmugasamy et al., 2011). Similarly ipaH gene which is present in multiple copies on both the plasmid and chromosome of Shigella spp is considered to be a good and specific diagnostic tool for the detection of Shigella from clinical and food samples (Kenia et al., 2010; Thong et al., 2005). James et al. (2000) sequenced and compared the O antigen gene cluster with other Gram negative bacteria and it showed that the wbgZ gene was conserved for S. sonnei. WbgZ gene was used for the molecular cloning of S. sonnei (Zou et al., 2001) and there was no report where this gene is used for PCR based detection. Gene wzy encoding for O-antigen polymerase is highly specific for identification of individual O antigens in case of S. boydii (Jiang et al., 2004). All the four Shigella species (S. flexneri, S. sonnei, S. boydii and S. dysenteriae) could be identified correctly with this standardized multiplex PCR using species specific primers in case of S. flexneri (ipaH), S. boydii (wzy) and S. sonnei (wbgz). S. dysenteriae was not included in our study. The standardized multiplex PCR has Shigella genus specific primer (ipaH1) which was helpful in identifying S. dysenteriae where amplification of the ipaH1 gene and IAC and absence of other amplicons confirmed the presence of S. dysenteriae. The selectivity and specificity of inv A gene for Salmonella enterica and ipaH1 gene for Shigella genus, as observed in our study confirms the earlier observation of Jasmid et al. (2008).
The multiplex PCR had a reasonably high level of sensitivity in experimentally spiked chicken samples. Detection of very low levels of bacterial contamination in food necessitates that these samples to be cultured for a few hours in peptone water for providing conditions for growth and multiplication of bacterial pathogens to a detectable level, dilution of inhibitory substances present in food and dilution of dead target cells, which provides some assurance that the detected DNA belongs from viable target cells. The multiplex PCR reported in our study had a reasonably high level of sensitivity in experimentally spiked chicken pulav samples and able to detect as low as 101 and 102 organisms per ml of Shigella and Salmonella enterica respectively following 5 h enrichment in peptone water thus the sensitivity was much better when compared to other reports (Babu et al., 2013; Ojha et al., 2013). According to Ohja et al. (2013) detection limit of mpcr after preincubation in GNB (Gram Negative Broth) was 5 x 104 cells and according to Babu et al. (2013) mpcr detection limit was 10 cfu /PCR after 10 h of incubation in BHI broth.
Epidemiological studies on Shigella have established that 10 cells are sufficient to be an infective dose (Wachsmuth and Morris, 1989). The results presented in this work showed that PCR with ipaH1 primers could be a useful tool for the detection as its sensitivity is better when compared with the reported multiplex PCR (Vantarakis et al., 2000). This detection sensitivity was adequate enough to precisely pickup the presence of these pathogens from among the natural food samples. However, the detection sensitivity of spiked food samples is higher when compared with pure reference culture which may be due to the overnight enrichment of spiked food samples. When evaluated on a few naturally occurring food and environmental samples, the multiplex PCR detected three samples positive for Salmonella, one sample positive for S. flexneri with identical results obtained following the conventional culture, isolation and biochemical identification procedures. This strengthens the claim of the developed multiplex PCR as a viable and reliable alternative for simultaneous detection of these organisms within 5-8 h when compared to the weeklong cumbersome isolation and identification procedures. Considering the low cost involved and relatively much shorter time needed to detect these important organisms this tool is useful for investigation of food borne outbreaks where these organisms are involved. Both multiplex PCR and conventional isolation and identification method showed good analytical and diagnostic accuracy. This means that non-target microorganisms tested were negative both by PCR and culture while all Salmonella and Shigella strains were positive by both methods. The multiplex PCR assay established in our study highlights the usefulness of the multiplex PCR for concurrent and rapid detection of Salmonella enterica and differentiation of Shigella spp from food and environmental samples.
Conclusion
The major contribution of this study was to develop a multiplex PCR to differentiate Salmonella enterica and Shigella spp. from other closely related members of Enterobacteriaceae family. The standardized multiplex PCR resulted in appropriate PCR amplicons while IAC alone amplified from non Salmonella and non Shigella species and in negative control where no template DNA was added. This protocol would help in detecting Salmonella enterica and Shigella genus and also differentiating Shigella spp., during routine investigations of food and clinical samples and also during the biological emergencies. The reported multiplex PCR in our study is simple, inexpensive and sensitive and enables the quick and precise detection of Salmonella and Shigella strains and could be used in the routine diagnostic laboratory.
Acknowledgments
Authors are thankful for DRDO for providing necessary facilities to carry out this work.
Submitted: January 9, 2013
Approved: September 9, 2013.
All the content of the journal, except where otherwise noted, is licensed under a Creative Commons License CC BY-NC.
- Alejandro G, Maria-Jose C, Belen R, Martina F, Jorge L, Juan MV, Ana GC (2012) Development of a multiplex real-time PCR method for simultaneous detection of Salmonella enterica, Shigella flexneri and Listeria monocytogenes in processed food samples. Eur Food Res Technol 234:571-580.
- Babu L, Reddy P, Murali HS, Batra HV (2013) Optimization and evaluation of a multiplex PCR for simultaneous detection of prominent food borne pathogens of Enterobacteriaceae. Ann Microbiol 63 (1). DOI 10.1007/s13213-013-0622-0.
- Batt CA (1997) Molecular diagnostic for diary based pathogens. J. Dairy Sci. 80:220-229.
- Brasher CW, De - Paola A, Jones DD, Bej AK (1998) Detection of microbial pathogens in shellfish with multiplex PCR. Curr Microbiol 37:101-107.
- Brigittle D, Phillppe JS, Parsof C (1998) Induction of Type 111 secretion in S. flexneri is associated with differential control of transcription of gene encoding secreted proteins. The EMB Journals 17:2894-2901.
- D'Aoust JY, (1989) Salmonella In: Doyle, M.P.(ed). Food borne bacterial pathogens. Marcel Dekker, Inc., New York, Bacel, pp 327-445.
- Farfan MJ, Garay TA, Prado CA, Filliol I, Ullao MT, Toro CS (2010) A new multiplex PCR for differential identification of Shigella flexneri and Shigella sonnei and detection of Shigella virulence determinants. Epidemiology and Infec 138:525-533.
- Food and Drug Administration. 1995. Bacteriological Analytical Manual. 8th ed. Association of Analytical Chemists. Arlington, VA, USA. Available at: http://www.fda.gov/Food/FoodScienceResearch/ucm70149.htm Accessed 12 December 2012.
- Germani Y, Sansonetti PJ, (2006) The genus Shigella In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt, E (eds) The Prokaryotes. A hand Book of the Biology of Bacteria. New York, pp 99-116.
- Hill WE (1996) The polymerase chain reaction for the detection of food borne pathogens. Crit.Rev.Food 36:123-173.
- Hill WE, Ferreira JL, Jones VM (1985) Probability of recovering pathogenic Escherichia coli from foods. Appl Environ Microbiol 49:1374-1378.
- Hoorfar J, Malorny B, Abdulmawjood A, Cook N, Wagner M, Fach P (2004) Practical considerations in design of internal amplification controls for diagnostic PCR assays. J Clin Microbiol 5:1863-1868.
- James GS, Wang L, Reeves PR (2000) Comparison of O-Antigen Gene Clusters of Escherichia coli (Shigella) Sonnei and Plesiomonas shigelloides O17: Sonnei gained its current plasmid-borne O-antigen genes from P. shigelloides in a recent event. Infect Immun. 68:6056-6061.
- Jashmid A, Bassami MR, Afshari NS (2008) Identification of Salmonella spp., and Salmonella typhimurium by a multiplex PCR- based assay from poultry carracases in Marshad - Iran. Int J Vet Res 3:43-48.
- Jiang T, Feng Lu, Senchekova S, Yang J, Sashkov AS, Guo H, Zhao G, Knirel YA, Reeves P, Wang L (2004) Structural and genetic characterization of S.boydii Type 13 O antigen. J Bacteriol 186:383-392.
- Jousilahti P, Madkour SM, Ambrechts T, Sherwin E (1997) Diarrheal disease morbidity and home practices in Egypt. Public Health 111:5-10.
- Jun W Li, Xiu Q Shi, Fu H Chao, Xin W Wang, Jin L Zheng, Nong S (2004) A study on detecting and identifying enteric pathogens with PCR. Biomedical and Environ Sc 17:109-120.
- Kenia BJ, Clyde BM, Rosario A (2010) Detection of Shigella in lettuce by the use of rapid molecular assay with rapid sensitivity. Braz J Microbiol 41:993-1000.
- Kong RYC, Lee SKY, Law TWF, Law SHW, Wu RSS (2002) Rapid detection of six types of bacterial pathogens in marine waters by multiplex PCR. Water Res 36:2802-2812.
- Kumar A, Singh SK, Bajpai D (2010) Phenotypic and genotypic characterization of Shigella spp. with reference to its virulence genes and antibiogram analysis from river Narmada. Microbiol. Res 165:33-42.
- Kumar S, Balakrishna K, Batra HV (2006) Detection of Salmonella enterica serovar Typhi (S. typhi) by selective amplification of invA, viaB, fliC-d and prt genes by polymerase chain reaction in multiplex format. Lett Appl Microbiol 42:149-154.
- Lantz PG, Hahnhagerdal B, Radstorm P (1994) Sample preparation methods in PCR based detection of food pathogens. Trends in Food Sc. Technol 5:384-389.
- Li Y, Mustafa A (2004) Simultaneous Detection of Escherichia coli O157:H7, Salmonella and Shigella in Apple Cider and Produce by a Multiplex PCR. J Food Protection 67:27-33.
- Li Y, Zhuang S, Mustaffa A (2005) Application of a multiplex PCR for the simultaneous detection of Escherichia coli O157:H7, Salmonella and Shigella in raw and ready-to-eat meat products. J Meat Sc 71:402-406.
- Liu B, Knirel YA, Feng L, Perepelov AV (2008) Structure and genetics of Shigella O antigens. FEMS Microbiol Rev 32:627-653.
- Mahesh S, Radhika M, Murali HS, Batra HV (2009) Detection of Salmonella enterica serovar Typhimurium by selective amplification of fliC, fliB, iroB, invA, rfbJ, STM 2755, STM 4497 genes by polymerase chain reaction in monoplex and multiplex format. World J Microbiol Biotechnol 25:1385-1394.
- Malorny B, Hoorfar J, Bunge C, Helmunth R (2003) Multicenter validation of the analytical accuracy of Salmonella PCR: towards an international standard. Appl Environ Microbiol 69:290-296.
- Miescier JJH, Redman DA, Salinger JA, Lucas JP, (1992) Molluscan shellfish: oyster, mussels and clams. In. Vanderzant, C., Splittstoesser. (eds). Compendium of Methods for the Microbiology Examination of Goods. Washington DC, American Public Health Association, pp 897-918.
- Moffat RMC, Appulamy R, Koye A, Carswell A, Denohy D (2012) An outbreak of Salmonella typhimurium Phage 135a gastroenteritis's linked to eggs served at an Australian Capital Territory cafe. Communicable Disease Intelligence 36:212-218.
- Niyogi SK (2005) Shigellosis. J Microbiol 43:133-143.
- Ojha SS, Yean CY, Ismail A, Singh KKB (2013) A pentaplex assay for the detection and differentiation of Shigella spp. Biomed Res International. Article ID 412370, 9 pages, 2013. doi:10.1155/2013/412370
- Olsen JE, Aabo S, Hill W (1995) Probes and polymerase chain reaction for detection of food borne pathogens. Int J Food Microbiol 28:1-78.
- Radji M, Malik A, Widyasmara A (2010) Rapid detection of Salmonella in food and beverage samples by polymerase chain reaction. Malayasian J Microbiol 6:166-170.
- Rahn K, Grandis SA-De, Clarke RC, Mcewen SA, Galan JE, Ginocchio C, Curtiss R, Gyles CL (1992) Amplification of an invA gene sequence of Salmonella typhimurium by polymerase chain reaction as a specific method of detection of Salmonella. Mol Cellular Probes 6:271-279.
- Shanmugasamy M, Velayutham T, Rajeswar J (2011) InvA gene specific PCR for detection of Salmonella from boilers. Vet World 4:562-564.
- Soumet C, Ermel G, Rose N, Drovin P, Salvat G, Colin P (1999) Evaluation of multiplex PCR for simultaneous identification of Salmonella spp, Salmonella enteritidis, and Salmonella typhimurium from environment swabs of poultry houses. Appl Environ Microbiol 28:113-117.
- Tao J, Feng Lu, Sennchekova SN, Shaskov AS (2004) Structural and genetic characterization of S.boydii Type 13 O Antigen. J Bacteriol 186:383-392.
- Theron J, Morar D, Preez DU, Brozel MVS, Venter SN (2001) A sensitive seminested PCR method for the detection of Shigella in spiked environmental water samples. Water Res 35:869-874.
- Thong KL, Hoe SLL, Puthucheary SD, Yasin RM (2005) Detection of virulence genes in Malaysian Shigella species by multiplex PCR assay. BMC 5. Available at http://www.biomedcentral.com/1471-2334/5/8
- Vantarakis G, Komninou DV, Papapetropoulou M (2000) Development of a multiplex PCR detection of Salmonella spp., and Shigella spp., in mussels. Lett App Microbiol 31:105-109.
- Villalobo E, Torres A (1998) PCR for detection of Shigella spp. in mayonisse. Appl Environ Microbiol 64:1242-1245.
- Wachsmuth K, Morris GK (1989) Shigella. In: Doyle, M.P. (ed) Food Borne Bacterial Pathogens. Marcel Dekker Inc., New York, pp 448-660.
- Wallace HA, Thomas SH (1998) Bacteriological Analytical Manual. 8th edition, Revision A, Chapter 5, www.cfsan.fda.gov/nebam/bam-5
- Way JS, Josephson KL, Pillai SD, Abbaszadegan M, Gerba CP, Pepper IL (1993) Specific detection of Salmonella spp. by multiplex polymerase chain reaction. Appl Environ Microbiol 59:1473-1479.
- Zou DQ, John OC, Ambulos N, Burr DH, Kopecko DJ (2001) Molecular cloning and characterization of genes from S. sonnei from I O-polysaccharide: proposed biosynthetic pathway and stable expression in a Live Salmonella vaccine vector. Inf Immun 70:4414-4423.
Send correspondence to
Publication Dates
-
Publication in this collection
25 July 2014 -
Date of issue
June 2014
History
-
Received
09 Jan 2013 -
Accepted
09 Sept 2013